|
Legenda:
Kreslení molekul
BKchem je program pro kreslení organických molekul (podobně jako například ISIS Draw, ChemSketch nebo SketchEl). Umí vytvářet soubory ve formátech SVG, CML[20], MOL a dalších. Obsahuje klasické nástroje pro tvorbu cyklických i acyklických struktur s libovolnými heteroatomy, stereovazbami, náboji, radikály. Je vhodný i pro tvorbu reakčních schémat, jednoduchých obrázků a popisků, se vzorci lze otáčet i v prostoru, ačkoliv nejsou k dispozici žádné 3D modely. Lze nastavovat barvu a šířku čar, vlastnosti písma, zobrazení vodíků. Program umí počítat sumární vzorce, molekulové hmotnosti, zastoupení prvků, formální oxidační čísla všech prvků, dále umí najít aromatické struktury, vygenerovat SMILES[21] a InChI[22], zkontrolovat chemickou správnost struktury. Je možné mít otevřeno několik sloučenin najednou a přepínat mezi nimi pomocí kaskádového menu. Lze nadefinovat vlastní templáty, tedy struktury a funkční skupiny, které budou často používány. Umí nakreslit molekulu podle zadaného InChI nebo SMILES. Umí si sám stáhnout strukturu z internetové databáze podle zadaného systematického (nebo známějšího triviálního) názvu (jména sloučenin se zadávají v angličtině, tedy např.: benzene, heptanoic acid...). Program podporuje několik jazyků, mimojiné i češtinu.
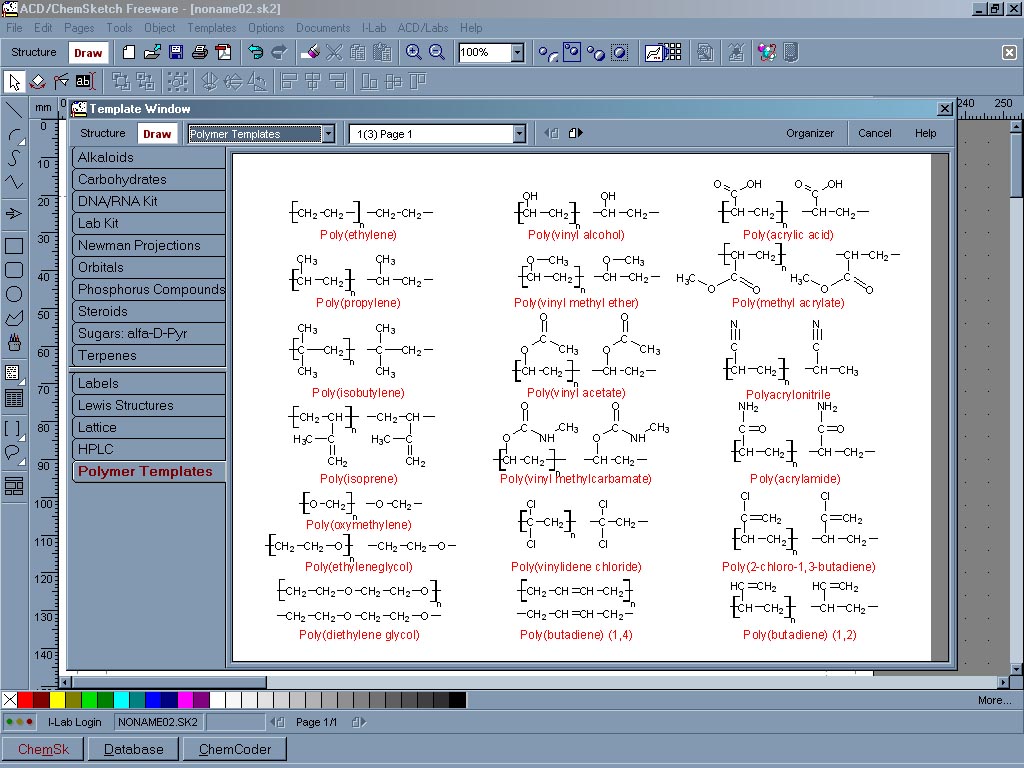
ChemSketch je propracovaný program pro tvorbu, úpravu a manipulaci s chemickými strukturami. Umožní nakreslit všechny běžné typy vzorců, makromolekul, reakčních schémat a chemických aparatur. Umí vytvořit delokalizované systémy, stereovazby, přehledná reakční schémata, automaticky očíslovat a popsat atomy. Pro usnadnění práce obsahuje vestavěné knihovny tematicky rozdělených templátů jako jsou alkaloidy, steroidy, terpeny, heterocykly, aminokyseliny, bicykly, karoteny, crownethery, sacharidy, fullereny, polymery, vitamíny a další. Dále tvary nejběžnějších orbitalů, krystalických mřížek, laboratorního nádobí a aparatur. S molekulou je možno pracovat v trojrozměrném prostoru a také ji naimportovat do sesterské aplikace 3D viewer, která je součástí ChemSketche. Program umí vygenerovat systematické názvy nepříliš složitých struktur, stereodeskriptory chirálních center a dvojných vazeb, InChI či SMILES. Umí odhadnout některé fyzikální a chemické vlastnosti nakreslených látek. Vyhledá tautomerní formy nakreslené látky, pokud existují. Pro účely publikování program obsahuje šablony pro úpravu nakreslených struktur dle požadavků jednotlivých časopisů. Může být použit i jako jednoduchý kreslicí program pro tvorbu tabulek, grafů a jednodušších obrázků. Program ChemSketch je freeware, ale pro jeho stažení je nutná registrace na stránkách ACD/Labs.
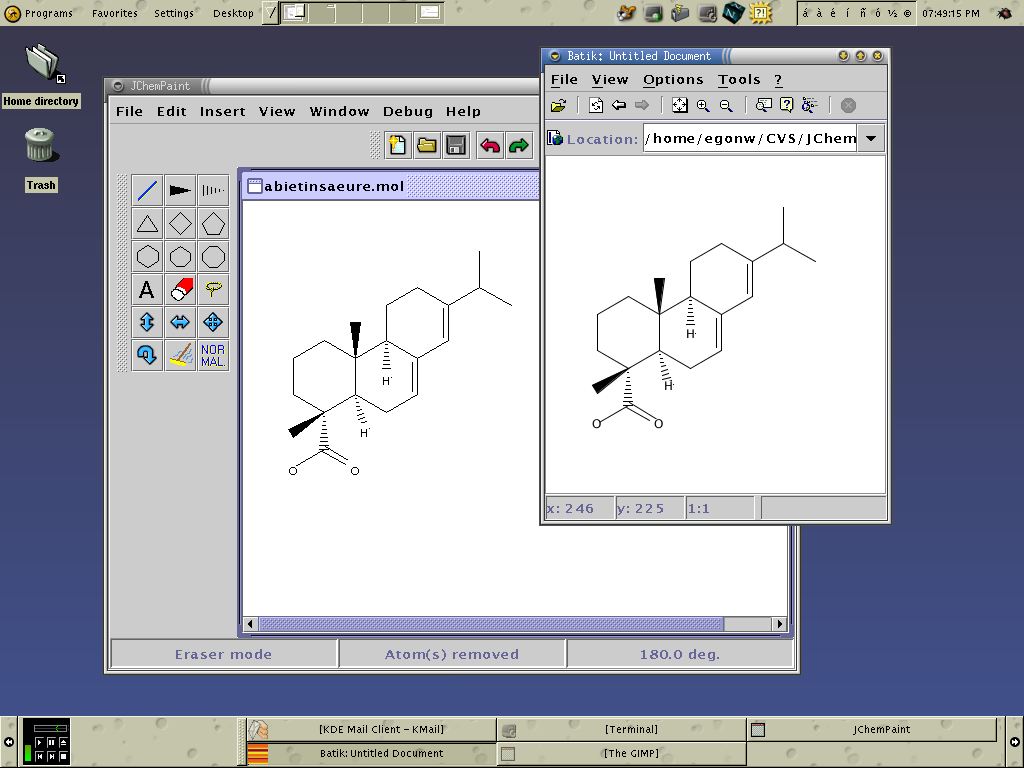
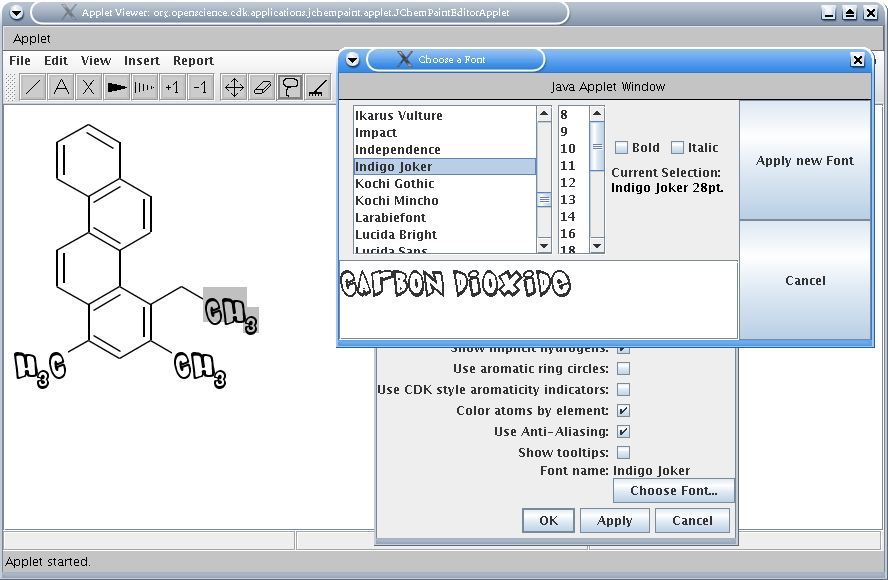
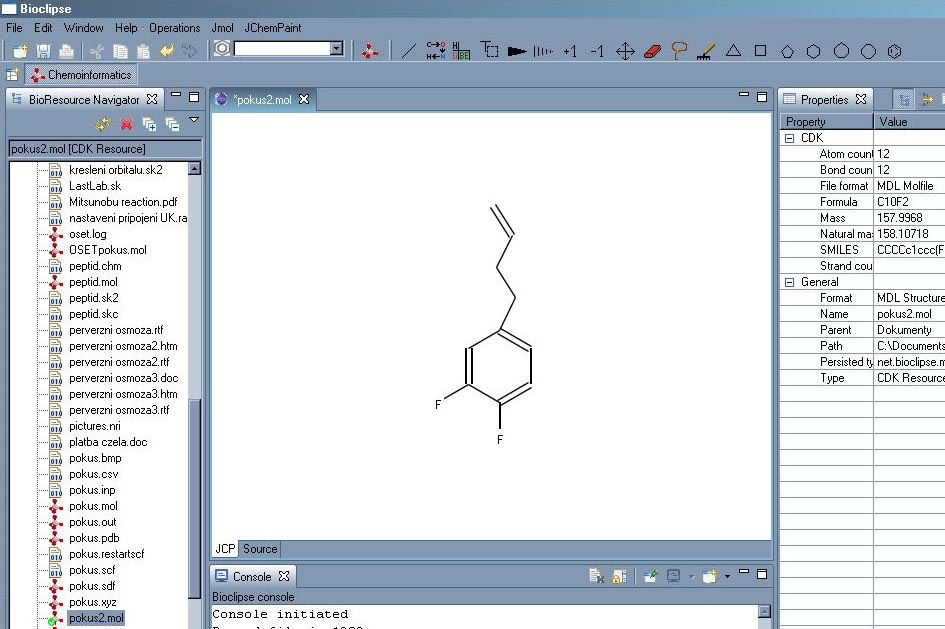
JChemPaint je editor pro kreslení molekul ve 2D. Je kompatibilní s programem JMol, který je určen pro zacházení s 3D strukturami. Oba tyto programy jsou napsány v Javě, pro jejich správný chod je třeba mít nainstalovaný JRE[a]. JChemPaint má samozřejmě základní funkce jako kreslení jednoduchých, násobných vazeb a stereovazeb. Umí rozpoznat aromatické systémy a kreslit delokalizované vazby. Dokáže vyexportovat nakreslenou strukturu jako obrázek ve formátu BMP, TIF, GIF, PNG, SVG nebo JPEG. Nakreslí strukturu podle zadaného kódu SMILES. Dokáže dle zadaného CAS-RN stáhnout strukturu sloučeniny z internetové databáze DOC (Dictionary on Organic Chemistry). Předpoví (v hrubé aproximaci) chemické posuny signálů C-NMR zadané látky.
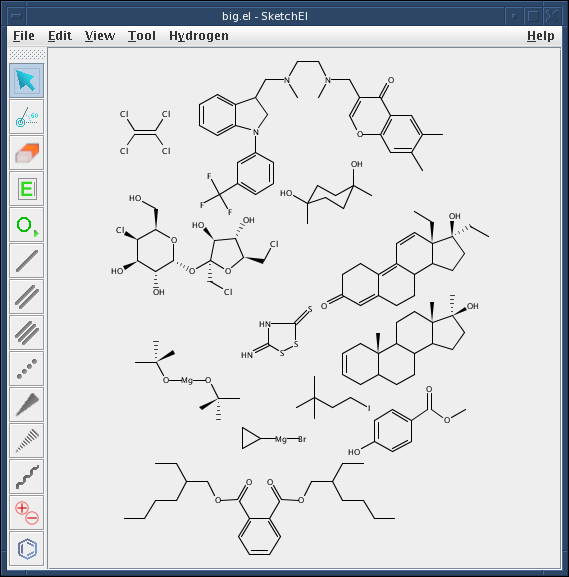
SketchEl je program napsaný v jazyce Java sloužící ke kreslení chemických struktur. Obsahuje běžné nástroje známé z aplikací podobného typu, které umožní nakreslit všechny běžné vzorce včetně stereochemických vazeb a nábojů. Obsahuje i malý soubor templátů některých nejběžnějších cyklických struktur. Struktury je možné libovolně zvětšovat a otáčet, zvolit jestli mají být vykresleny vodíkové atomy apod. Je možné vygenerovat tzv. Z-matici, tedy soubor všech atomů dané sloučeniny s jednotlivými souřadnicemi a náboji každého elementu. Obrázky lze z programu exportovat ve formátu MOL a nebo jednodušeji použitím klasického copy-paste. Neumožňuje bohužel tvorbu reakčních schémat a sofistikovanějších trojrozměrných obrázků. Aplikace je vhodná pro rychlé a jednoduché kreslení nepříliš komplikovaných struktur. Program potřebuje nainstalované rozhraní JRE.
WinDrawChem je další z řady editorů určených pro kreslení 2D chemických stuktur. K dispozici jsou všechny klasické nástroje, řada templátů pro snadnější tvorbu cyklických struktur, aminokyselin, nuklových kyselin, sacharidů, chránících skupin. Templáty si lze nadefinovat i dle vlastní podle potřeby. WinDrawChem do sebe má implementovanou aplikaci OpenBabel, takže dokáže exportovat i importovat vpodstatě všechny běžné chemické formáty. Obrázky lze ukládat například ve formátu BMP či PNG a nebo jednodušeji použít klasické funkce copy-paste. Samozřejmostí je možnost upravit vzhled molekul (velikost, barva, písmo). Lze kreslit buď normalizovaně (vazby mají definované délky a úhly) nebo libovolně. Automatické nastavení lze upravovat. Program umí spočíst sumární vzorec, molekulovou hmotnost, elementární analýzu. Součástí je i jednoduchý prediktor H-NMR a C-NMR spekter (chemické posuny a multiplicity) a IR spekter (vlnočet a typ pásu). Je možné kreslit i reakční schémata, program umí spočítat změnu enthalpie reakce a porovnat NMR spektra reaktantů a produktů. Jsou podporovány SMILES. 3D Vizualizace
Bioclipse je program pro vizualizaci a úpravy chemických struktur. Je to souborový manažer, editor a 3D prohlížeč v jednom. Je to Java aplikace, zahrnuje v sobě několik pluginů jako například dříve diskutovaný JChemPaint nebo Jmol a další mohou být lehce implementovány. Struktura se současně zobrazí v editačním a 3D okně, v dalších jsou pak nejrůznější dostupné informace o molekule (sumární vzorec, složení, SMILES notace a další).
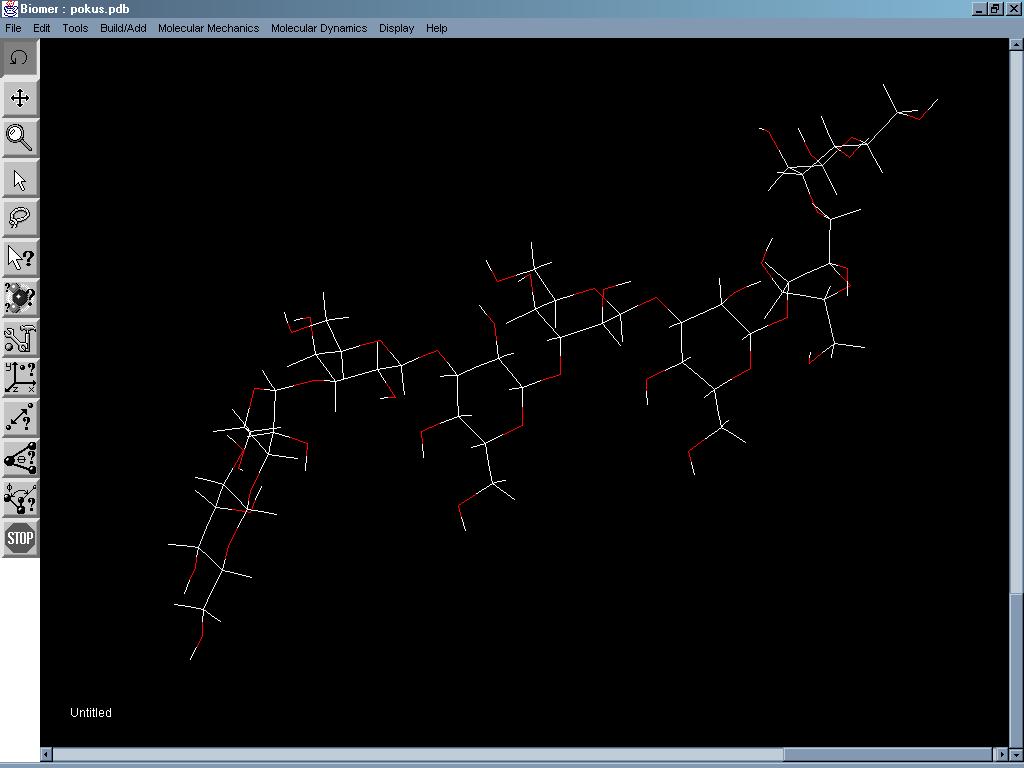
Biomer (někdy také označovaný jen jako B) je program určený pro biomolekulární modelování. Není příliš vhodný pro přímé kreslení menších molekul, ale umí je bez problému načíst ve formátu PDB. Tyto je už potom poměrně snadné editovat. Program obsahuje templáty obvyklých funkčních skupin, kruhů, alifatických zbytků a podobně. Naopak snadné je vytváření řetězců peptidů, polysacharidů a polynukleotidů. Nadefinuje se pouze pořadí jednotlivých aminokyselin a určí se konformace řetězce (popřípadě pořadí monosacharidů, konektivita, L/D isomerie a druh anomeru, respektive pořadí bazí a konformace - u polysacharidových řetězců, respektive DNA/RNA). Vygenerované struktury lze poté podrobit dalším úpravám, například pomocí výše zmíněných, v programu již připravených zbytků. Pro zobrazení struktury je k dispozici několik druhů různých modelů. Program spočítá molekulovou hmotnost a náboj struktury, počítá vzdálenosti, úhly a torsní úhly, zjistí přibližnou energii dané struktury, z chirální látky umí vytvořit její enantiomer, také umí optimalizovat geometrii molekuly (jednoduchou semiempirickou metodou) nebo simulovat vibrace při zadané teplotě. Je třeba mít nainstalované JDK a JRE.
Program podporuje velké množství běžných chemických formátů souborů. Po načtení struktury se zobrazí její 3D model se kterým lze snadno manipulovat (levé tlačítko myši zajišťuje otáčení v prostoru, prostřední translaci a pravé mění velikost). Pro zpřehlednění složitějších struktur (polysacharidy, proteiny..) lze zobrazit pouze vybrané části molekuly (což také značně urychlí manipulaci se strukturou, zvláště na pomalejších počítačích). K dispozici jsou různé modely zobrazení (wire,ball&stick,sphere) v široké paletě barev. Nastavení lze aplikovat na celou strukturu či jen na určitý podřetězec, určité zbytky nebo atomy. Do celého modelu či jen na vybraná místa je možné nechat automaticky doplnit značky nebo názvy atomů a funkčních skupin, v případě proteinů i názvy aminokyselinových zbytků. Struktury mnoha proteinů si lze zdarma obstarat z internetové databáze[23], v programu jen stačí zadat identifikační kód sloučeniny a Chimera si ji sama stáhne. Rozdělaná práce se ukládá jako soubory formátu PY, obrázky se exportují například jako JPEG nebo PNG. Je možné vytvářet i animace ve formátu MOV, MPEG, MP4. Program Chimera lze ovládat v zásadě dvěma způsoby. Myší přes klasická rolovací menu nebo příkazovou řádkou. První způsob vyžaduje obecně více kroků, ale není třeba si pamatovat jednotlivé příkazy. Ovládání pomocí textových příkazů je ovšem rychlejší a elegantnější.
CueMol je program pro
vizualizaci makromolekul, zejména proteinů. Zvládá několik
různých modelů, umí zobrazit mapy elektronových hustot, počítat
vzdálenosti a úhly mezi atomy. Program lze ovládat i přes CLI.
DeepView je nástroj
vhodný pro analýzu a zobrazení makromolekul, obzvláště proteinů,
původně byl vyvinut pro použití spolu s výpočetními nástroji
ExPASy[24].
Program načte soubory PDB a MOL, popřípadě je peptid
vytvořen přímo v programu prostým zadáním sekvence aminokyselin. Umí
najít potencionální vodíkové můstky uvnitř proteinu i mezi proteinem
a ligandem, načíst mapy elektronových hustot z krystalografických
experimentů. Umí odhadnout strukturu zadaného proteinu tak, že ji odešle
na ExPASy server, kde je porovnána s jí podobnými strukturami v databázi.
Discovery Studio je software pro zobrazování, modelování a simulaci vlastností chemických látek. Podporuje většinu běžných chemických formátů souborů, 2D struktury jsou po importování automaticky převedeny na trojrozměrné, lze je zobrazit s použitím běžných modelů (wireframe, ball&stick..), zvolit barvy a rozlišení podle potřeby. Na centrech chirality je možné zvolit požadovanou konfiguraci, pro lepší přehlednost skrýt nepodstatné skupiny a řetězce, nechat molekulu v prostoru volně otáčet. Program je vhodný zvláště pro práci s velkými molekulami (proteiny, DNA...), obsahuje mnoho funkcí pro zacházení právě s látkami tohoto typu. Data lze ukládat v mnoha formátech, lze exportovat obrázky nebo VRML soubory pro implementaci do webových stránek. Může být otevřeno několik struktur najednou a pak mezi nimi přepínat pomocí kaskádového menu. Složitější struktury jsou zobrazeny ve dvou oknech současně: v tzv. 3D okně je zobrazena ve vysokém rozlišení samotná látka a tzv. sekvenčním okně se nacházejí informace o její struktuře, pořadí monomerů apod. Program umí mimojiné vytvářet animace, pracovat s krystalovými mřížkami, namapovat povrch s konstantním elektrostatickým potenciálem, simulovat přístupnost jednotlivých částí molekuly rozpouštědlu. Program je freeware, ale před jeho používáním je nutné se zaregistrovat na www.accelrys.com. Nutná je bohužel registrace i při přístupu k tutorialům nebo dalším oficiálním zdrojům na webu.
gOpenMol je program pro zobrazení a analýzu molekul a jejich vlastností, které byly předtím spočteny v nějakém externím výpočetním programu (Gaussian, Gamess, Jaguar, Spartan...). Lze ho ovládat buď pomocí grafického prostředí nebo příkazové řádky. Grafické prostředí gOpenMolu se skládá z grafického a příkazového okna. V grafickém okně se zobrazí importovaná struktura, jejíž pohyb se ovládá myší (rotace, translace, změny velikosti). Je možné měřit vzdálenosti, úhly a torsní úhly mezi vybranými atomy. Struktury je možné editovat, mazat a přidávat atomy či upravovat konektivitu. V principu je možné naimportovat jakoukoliv molekulu v podporovaném formátu, některé funkce jsou ale samozřejmě dostupné jen pro výstupní soubory z výpočetních programů. gOpenMol umí použít výstupní data například k zobrazení molekulových orbitalů, vykreslení elektronových hustot, namapování elektrostatického potenciálu, zobrazení RDF. gOpenMol pracuje s nepříliš rozšířenými soubory typu PLT, ale sám si umí do tohoto formátu převést soubory typu MAP, IRC, OUT, LOG a další. Struktury lze tradičně zobrazit s pomocí několika typů modelů (wire, stick, sphere), lze upravovat 'osvětlení' a 'materiál' modelu pro vytváření různých 3D efektů, upravovat barvy, zobrazovat popisky atomů a zbytků, zobrazit souřadnice, náboje, vdW poloměry, konektivitu a molekulovou hmotnost všech atomů. gOpenMol umí vytvářet i animace. Obrázky struktur lze exportovat jako BMP, JPG, RGB, TGA soubory, zvlášť lze ukládat namapované povrchy nebo jen souřadnice atomů molekuly (CML, PDB, XYZ...), všechna data se v průběhu práce ukládají do GOM (gOpenMol) souborů.
Slouží k prohlížení chemických struktur v 3D. Program je napsaný v Javě, k jeho chodu je třeba nainstalovat JRE. Může být začleněn jako komponenta do jiných Java aplikací. Umí načíst velkou škálu formátů souborů (buď nakreslených v chemickém editoru nebo výstupy výpočetních programů: Gaussian, Gamess, Jaguar, MOPAC, Ghemical, Spartan, Abinit). Strukturu lze zobrazit pomocí několika modelů (wireframe, spacefill, ball&stick...), lze libovolně upravit vzhled a barvy, nechat ji otáčet v prostoru, simulovat vibrace molekuly. Umí najít potenciální vodíkové můstky. Umí vytisknout strukturu nebo vyexportovat obrázky ve formátu JPG, PNG, PPM, podporuje PDF. Ze zobrazené struktury se dá vytvořit animace. Všechny úpravy operace mohou být prováděny buď klasicky pomocí grafického interaktivního rozhraní, nebo v konzoli jednoduchými příkazy RasMol/Chime Scriptů.
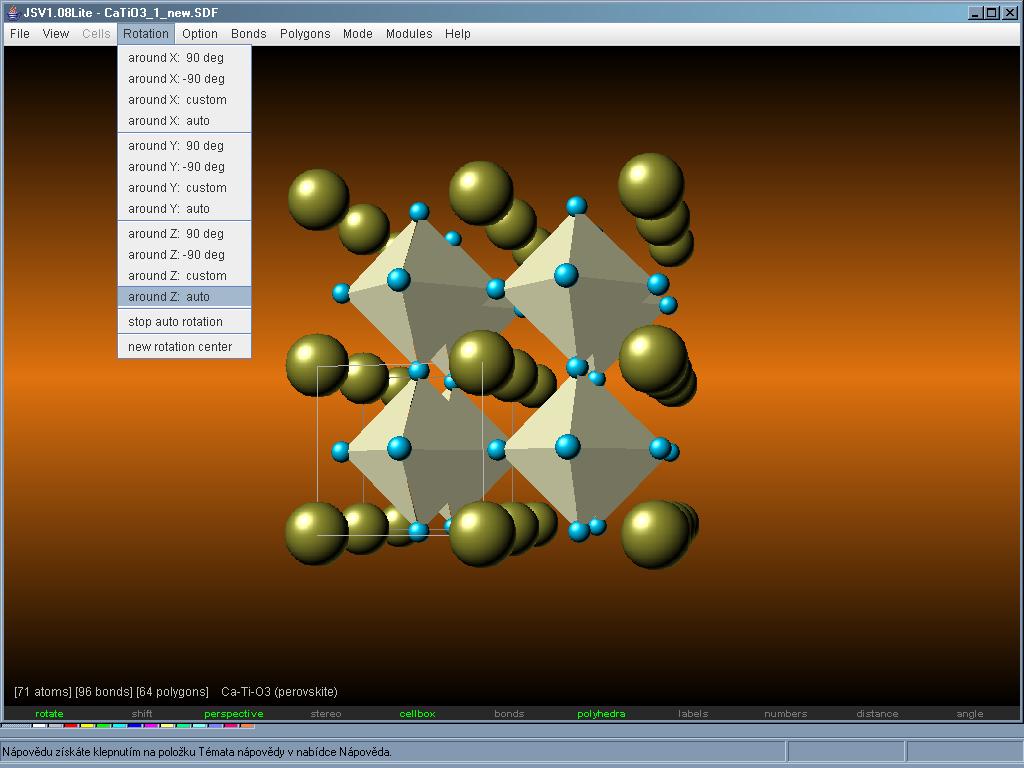
JSV nabízí tvorbu, editaci a vizualizaci krystalových struktur a mřížek, nanostruktur (nanotrubky, nanovodiče, fullereny) a podobně. Používá specifické JSV soubory (jedná se o čistě textové soubory) ale umí zacházet i s formáty SDF, PWD, CIF a CEL. V dané verzi bohužel ještě není implementována podopora CC1 (Fullerene Library) a PDB (Protein Data Bank) a vypadá to, že už ani v budoucnu nejspíš nebude. Program JSV tedy umí vytvořit a zobrazit danou strukturu, změřit vzdálenosti a úhly mezi atomy, spočíst mřížkové parametry, odhadnout hustotu látky, zjistit grupu symetrie, spočítat rentgenovou difrakci a další. JSV v sobě obsahuje několik dalších menších programů, které umožňují například další prohlížení vytvořené struktury, sledování zatížení paměti počítače a také zajímavou periodickou tabulku prvků.
Program pro vizualizaci krystalových struktur. Dokáže načíst strukturu
molekuly (v různých formátech), přepovědět a zobrazit, jak
bude vypadat její krystalická mřížka. Samozřejmostí je rotace a
translace v 3D a různé druhy vizualizace (ball and stick, wireframe,
capped sticks, spacefill). Zobrazí krystalové mřížky a základní
buňky, najde a zobrazí možné intra- a intermolekulární vodíkové
můstky i další vybrané nevazebné interakce. Umožní měřit délky
vazeb, vazebné a torzní úhly. Umí spočíst a zobrazit spektrum rentgenové
difrakce. Jak toto spektrum tak i krystalové struktury jdou uložit v mnoha
formátech včetně JPG a BMP.
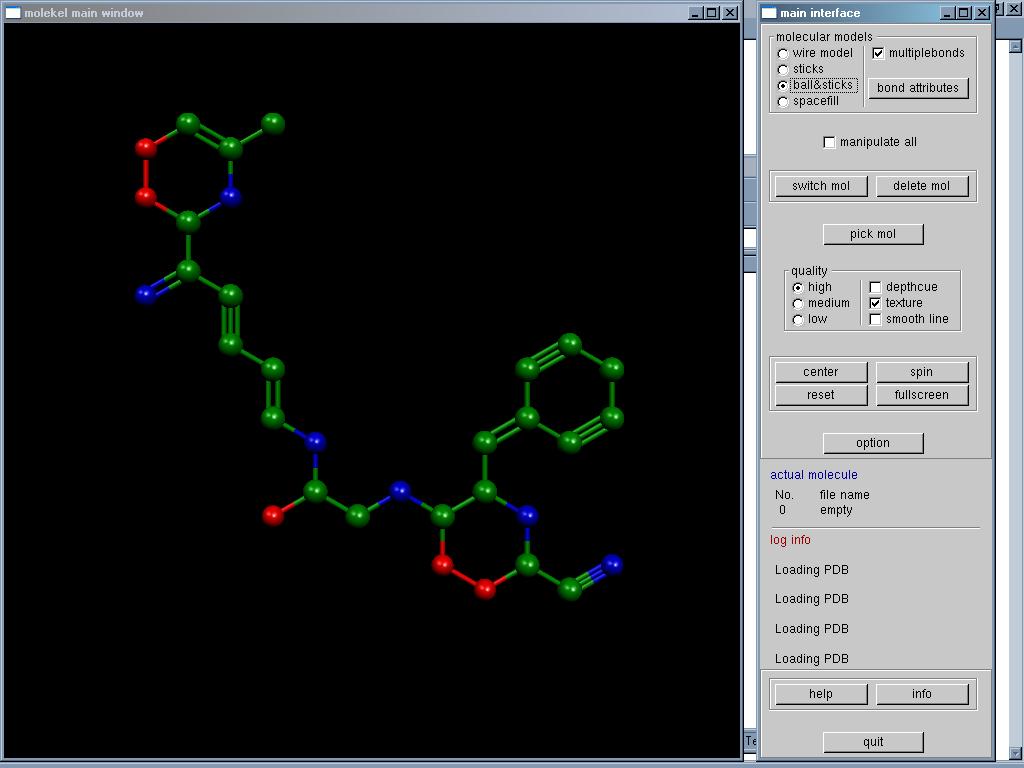
Molda je nástroj pro
kreslení a 3D vizualizaci molekul. Může sloužit i jako interface k
některým výpočetním programům (MOPAC, Gaussian...).
Program je jednoduchý a
přehledný, jeho vývoj byl však před třemi lety zastaven,
nemůže se tedy po grafické stránce rovnat dalšímu software ze své
kategorie. Existuje i Java klon Moldy, pod jménem Molda for Protein
Modelling.
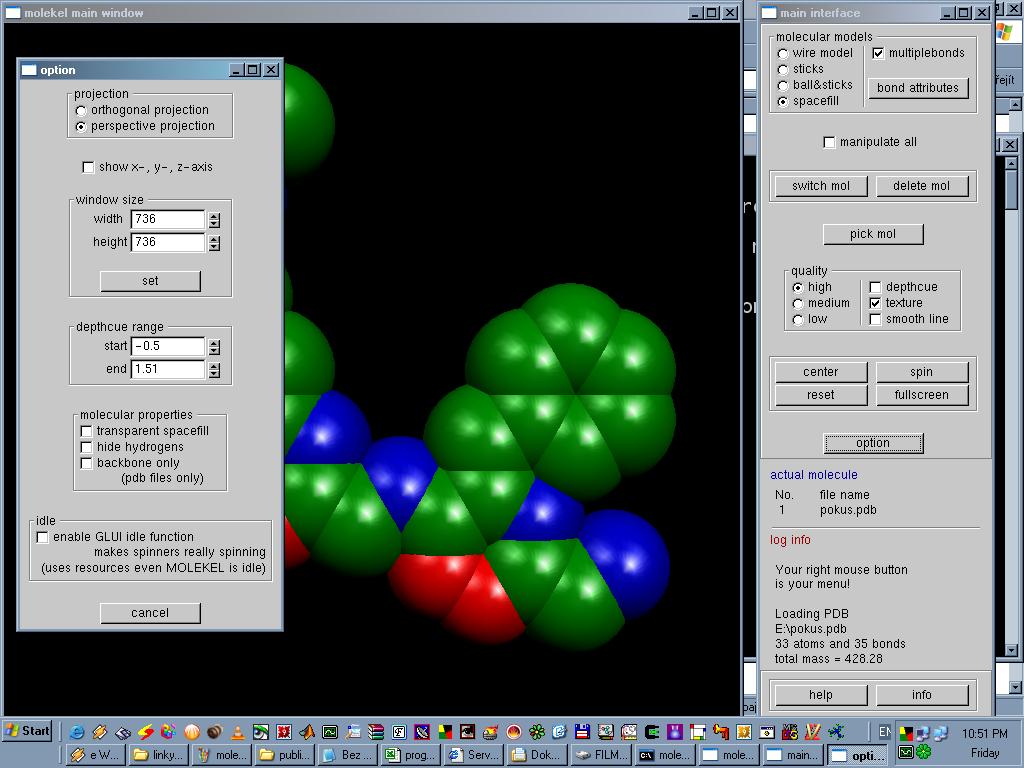
Grafické rozhraní programu Molekel se skládá ze dvou oken, tzv. grafického okna a hlavního okna. V hlavním okně je možné upravovat vzhled molekuly i celého programu, je zde také chronologický přehled všech operací a příkazů a informace o aktuálním otevřeném souboru. Kliknutím pravého tlačítka v grafickém okně se otevře ještě tzv. rolovací menu s nabídkou několika příkazů, z nichž určitě nejdůležitější je příkaz load (tedy načtení struktury), to je totiž právě to, co program Molekel dokáže: zobrazit molekulové struktury ve formátu XYZ, PDB, a hlavně výstupní soubory z výpočetních programů Gaussian a Gamess a dalších. Molekulu lze zobrazit jako drátěný model (wire), kalotový model (spacefill) popřípadě "stick" nebo "ball&stick" model; lze zvolit kvalitu zobrazení a nechat molekulu v okně pro lepší přehled libovolně automaticky otáčet. Strukturou je možné pohybovat, zvětšovat ji, zobrazit popisky všech atomů včetně nábojů. Je možné mít v jednom okně zobrazeno několik molekul najednou a zacházet s nimi jednotlivě nebo zvlášť. Program umí mimo jiné: změřit vzdálenost mezi dvěma požadovanými atomy, změřit úhel mezi libovolnými třemi atomy, změřit dihedrální úhel, spočíst dipólový moment, zobrazit energie a obsazení všech orbitalů (lze i uložit do samostatného souboru ve formátu MACU), zobrazit elektronové hustoty a elektrostatický potenciál, zobrazit vibrace molekuly, přehrát animaci průběhu optimalizace struktury. Všechny spočtené povrchy mohou být uloženy ve formátu PDB nebo XYZ. Obsah obrazovky lze v jakémkoliv okamžiku uložit jako obrázek (ve formátu JPG,RGB,TIF). Při používání výstupního souboru programu Gaussian (LOG, OUT) je pro možnost využití všech funkcí programu Molekel nutno v Gaussianu ještě před zahájením výpočtu zadat klíčová slova "gfprint" a "pop=full". Program je freeware, ale pro jeho stažení je nutná krátká registrace na jeho domovské stránce.
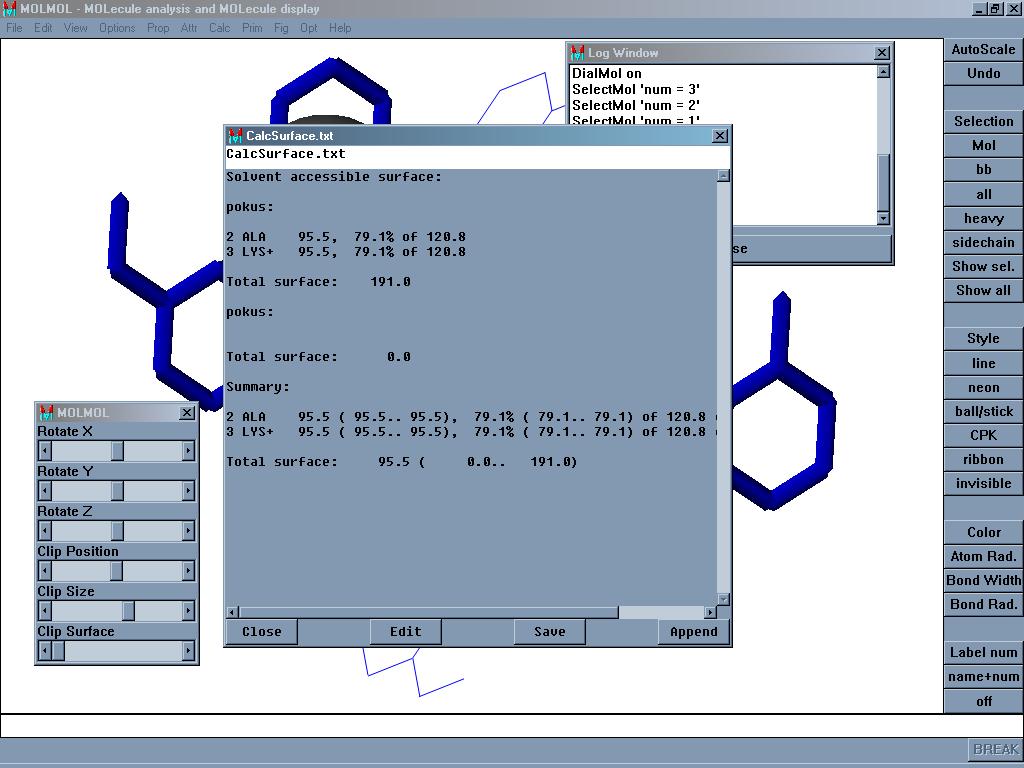
Molmol je program zobrazení a analýzu molekul a makromolekul. Načte a uloží soubory v mnoha formátech, dokáže spočítat vzdálenosti, úhly, potenciální vodíkové můstky, Ramachandranův graf (u peptidů) a další.
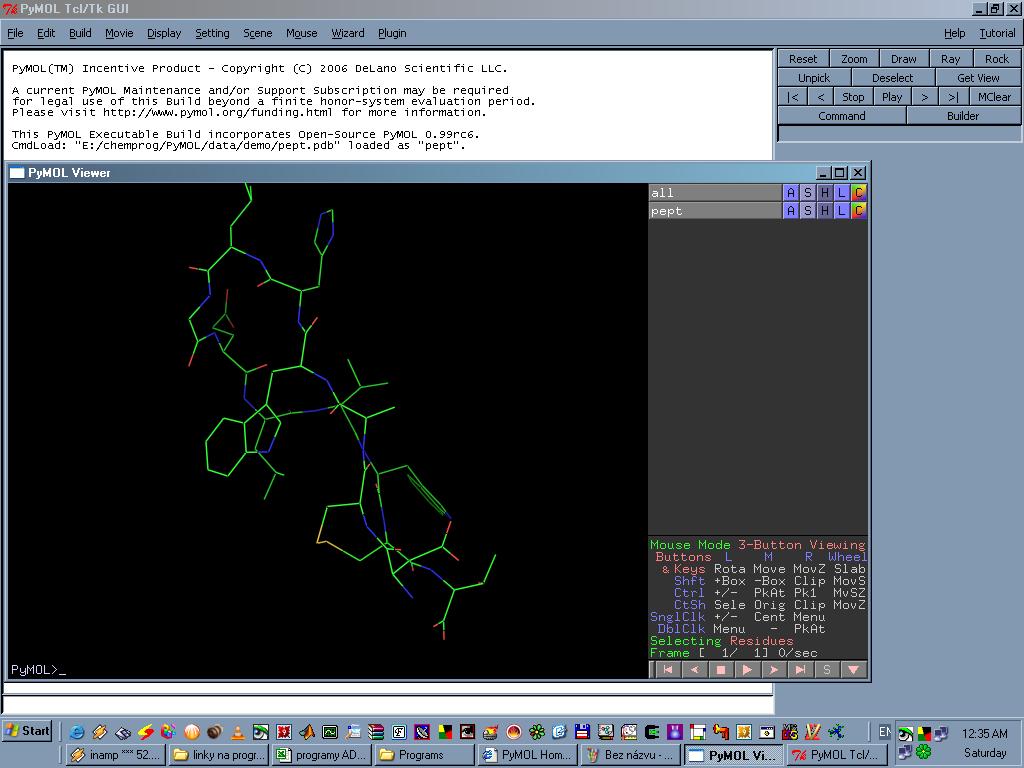
PyMOL je program pro
zobrazení a editaci především PDB, ale i moha dalších chemických
souborů. Dokáže renderovat obrázky i animace ve vysokém rozlišení, lze
vygenerovat vpodstatě jakýkoliv požadovaný druh zobrazení. Program se dá současně
ovládat myší a příkazovou řádkou.
Program pro zobrazení chemických struktur v 3D a vytváření animací. Dokáže načíst data ve formátu PDB (struktura může být nakreslena v nějakém běžném editoru, např. SketchEl a převedena v programu OpenBabel na PDB) nebo si je (pokud se jedná o známý protein) může automaticky stáhnout z databáze www.rcsb.org. Molekulu lze zobrazit pomocí několika modelů (wireframe, points, stick, ball&stick..), nastaví se kvalita rozlišení a množství 3D efektů, podle rychlosti počítače. Lze zjistit podobnost dvou molekul v RMSD[b]. Dále lze například hledat možné vodíkové můstky, měřit vzdálenosti a úhly, zobrazovat popisky atomů, vytvořit stereoobraz struktury, namapovat povrch struktury. V jednom okně může být zobrazený libovolný počet struktur, můžou být upravovány dohromady nebo zvlášť. Animace lze exportovat ve formátu avi.
RasMol je jednoduchý
program pro otevření souborů formátu PDB. Umí molekulu zobrazit s
pomocí libovolného modelu a vyexportovat ji jako obrázek nebo nechat
vytisknout. Jednoduchý malý program, neobsahuje žádné složitější funkce,
bývá někdy implementován jako grafické okno do nejrůznějších
aplikací.
RasTop vychází z
aplikace RasMol a nabízí uživateli i podobné možnosti. Má ale více
funkcí, propracovanější GUI a podporuje více vstupních formátů
souborů.
Program pro renderování obrázků s vysokou kvalitou a tvorbu animací.
Struktura musí být importována z nějakého externího programu ve formátu
PDB, MOL nebo XYZ. Program má velmi přehledné a intuitivní GUI, podporuje
angličtinu a italštinu.
VENUS je programový balík, napsaný v ANSCI C, pro zobrazování a manipulaci s krystalovými strukturami a mapami elektronových a jaderných hustot. Tyto hustoty mohou buď pocházet z experimentu rentgenové nebo neutronové difrakce a nebo z teoretického výpočtu z několika ab initio programů. Podporovány jsou komerční Gaussian, WIEN2k, SCAT a freeware GAMESS, VASP a ABINIT. VENUS se skládá celkem ze čtyř samostatných programů: VICS: (Visualization of Crystal Structures), VEND (Visualization of Electron/Nuclear Densities), PRIMA (PRactice Iterative MEM Analyses), ALBA (After Le Bail Analysis). První dva slouží k vizualizaci krystalových struktur a hustot, třetí pak ke spočtení elektronových resp. jaderných hustot z difrakčních dat. Program se skládá celkem ze tří grafických oken, jedno slouží k zobrazení struktury s použitím jednoho z pěti modelů a další k dvě k nastavení vlastností zobrazení a manipulaci s molekulou. Zdrojový kód není k dispozici, autoři chtějí program dále vyvíjet sami. K dispozici je rozsáhlá dokumentace.
Lze načíst jakkoli velkou molekulu ve formátu PDB, je i možné zadat jen kód příslušného proteinu, který má být zobrazen a program si ho sám stáhne z internetové databáze. Struktury nejsou omezeny ani velikostí, ani počtem atomů, jen pamětí počítače. Molekula se zobrazí ve zvláštním okně (tzv. OpenGL displej). Molekula může být zobrazena mnoha způsoby, k dispozici jsou desítky kombinací modelů a obarvení. Změny vzhledu mohou být aplikovány buď na celou molekulu, nebo jen na určitou část, lze vybrat atomy určitého druhu, kostru molekuly, postranní řetězce, k dispozici je mnoho možností, podle toho, k jakému účelu má animace sloužit a co má být zobrazeno. Pokud je importovaná molekula protein, lze zobrazit tabulku se sekvencí jeho aminokyselin, vybrané aminokyselinové zbytky se označí i v trojtozměrném modelu. Model může být v průběhu práce uložen jako VMD soubor, molekuly v OpenGL displeji lze ukládat jako BMP obrázky. Všechny operace prováděné s molekulou lze renderovat jako animace. Program se ovládá buď v klasickém GUI myší nebo jednoduchými příkazy v programové konzoli.
Výpočetní chemie
Abinit je balík
několika výpočetních programů. Jeho jádro, program Abinis,
zvládá obvyklé kvantové výpočty týkající se mimojiné optimalizace
geometrie, výpočtu celkové energie struktury a elektronových hustot. Jsou
používány DFT metody se zahrnutím některých korelací, například
pomocí výměnného-korelačního funkcionálu. Metodou TDDFT můžeme
spočítat například excitační energie či optická spektra.
Program tradičně postrádá GUI, spouští se z příkazové
řádky, kde je mu také zadána cesta ke vstupnímu souboru, který obsahuje
informace o počítané molekule a typu výpočtu. Vstupní soubory pro Abinit
jsou specifické, ale informace o jejich struktuře lze snadno získat na
stránkách programu, kde existuje rozáhlá dokumentace, která se mimojiné
věnuje jejich syntaxi. Snazší cestou je pak použítí některého z
vizualizačních nástrojů, které jsou vhodné i pro zobrazení výsledků.
Abinit je v tomto ohledu kompatibilní mimojiné s programy VASP Data
Viewer, Scilab, XCrysDen, OpenDX, Jmol, VENUS, XtalEdit, MOLDEN, Mercury.
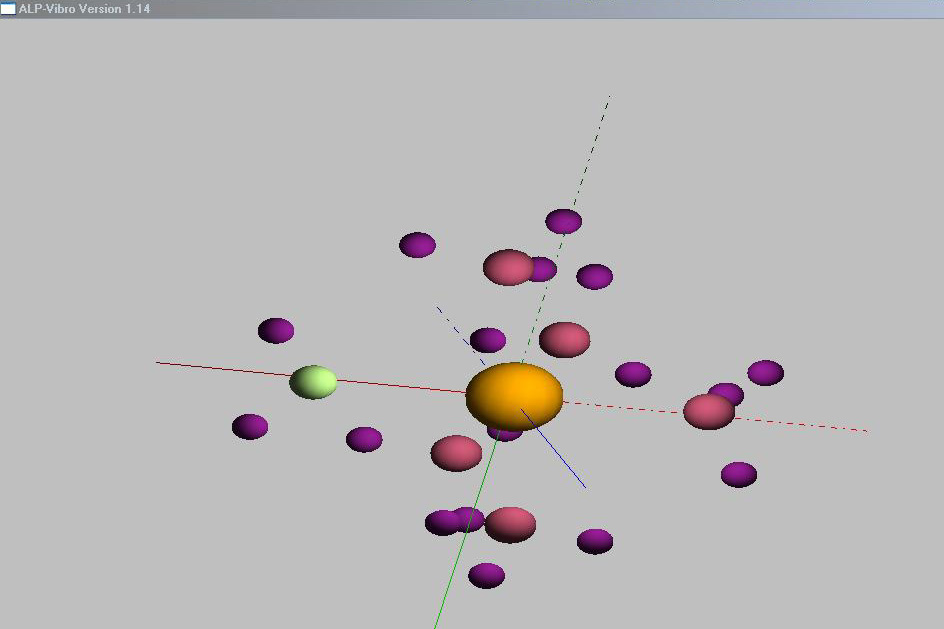
ALP-vibro je program
pro animaci vibrací molekuly za použití výstupu z programů GAMESS nebo
Gaussian. Jediná potřebná vstupní data jsou soubory typu OUT, animace
bohužel nelze ukládat.
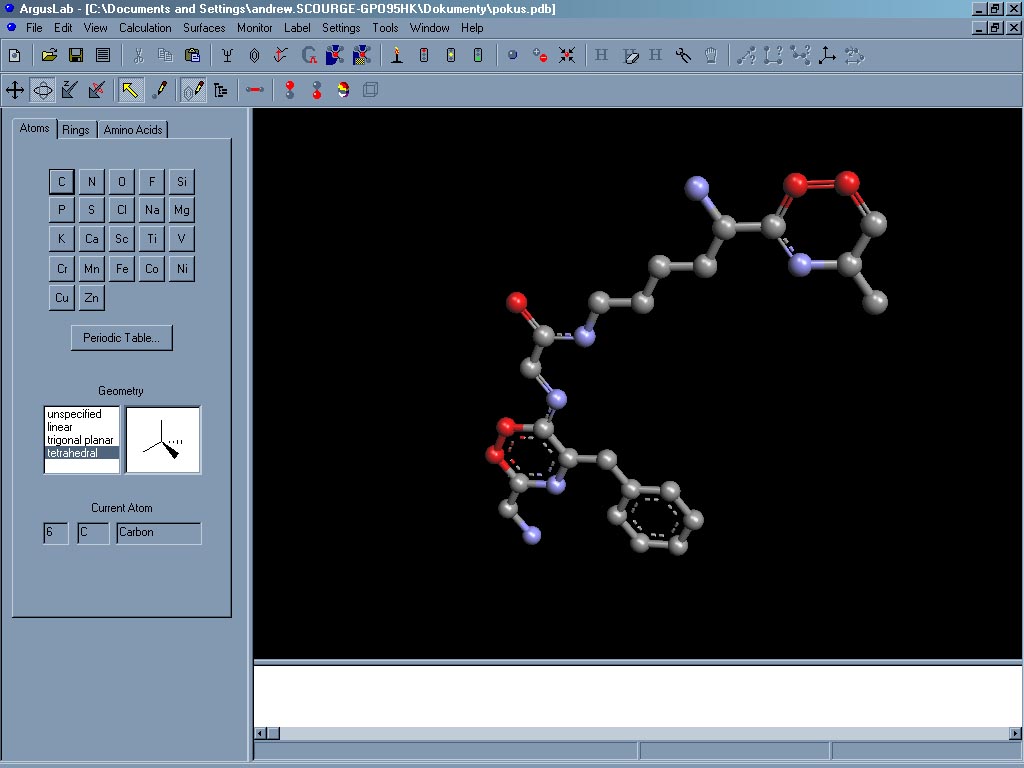
ArgusLab je další program z oblasti výpočetní chemie. Umožní načíst nebo vymodelovat požadovanou strukturu a poté provést optimalizaci její geometrie, spočíst energii dané fixované geometrie či spočítat její elektronová (UV/VIS) spektra, dipólové a magnetické momenty, odhadnout přístupnost jednotlivých částí molekuly rozpouštědlu. Zobrazí mapy elelektrostatického potenciálu či orbitaly (HOMO, LUMO). Program má přehledné grafické rozhraní, snadno a intuitivně se ovládá. Výpočty jsou prováděny semiempirickými metodami (AM1, PM3, MNDO), jsou velmi rychlé, výsledky jsou samozřejmě tomu odpovídající. ArgusLab ovšem může sloužit i jako vizualizační nástroj pro program Gaussian, umí vytvořit příslušný vstupní soubor a také ho Gaussianu rovnou odeslat. Výsledy lze uložit v několika běžných chemických formátech, lze exportovat obrázky. Program ArgusLab je freeware, ale pro jeho stažení je nutná registrace na stránkách http://www.arguslab.com.
Vizualizační nástroj pro výpočetní programy Gaussian, Gamess,
Molcas, Molpro a MPQC. Umožní nakreslit libovolnou strukturu ve 3D
nebo načíst její geometrii z některého z podporovaných formátů.
Na základě tohoto sestaví příslušnou Z-matici a umožní také nastavit
parametry výpočtu pro vlastní výpočetní program: metodu výpočtu
(Hartree-Fock, MP, DF...), bázi (STO-3G, 3-21G, 6-31G...), typ výpočtu
(optimalizace geometrie, energie dané geometrie) a další. Gabedit odešle
tato data příslušnému programu a po provedení výpočtu zobrazí
výsledky: molekulové orbitaly (HOMO, LUMO), plochy elektronových hustot, plochy
konstantního elektrostatického potenciálu (ESP), stínění magnetického pole
(pro NMR). Je možná editace jak vstupního textového souboru, ve kterém se
odesílají data daným ab initio programům, tak i prohlížení a editace
výstupu už během výpočtu (formát LOG a OUT). Gabedit umí zobrazit i
spočtená Ramanova či infračervená spektra dané látky. Všechny
obrázky mohou být uloženy v BMP, JPG, PNG, PPM nebo PS formátu. Také umí
automaticky vygenerovat sérii obrázků, ze kterých mohou být vytvořeny
animace (konvergence geometrie během výpočtu, vibrace a rotace
molekuly...).
Gamess (General Atomic and Molecular Electronic Structure System) je snad nejznámější freeware program pro kvantově chemické výpočty. Umí najít tranzitní stavy, optimalizovat geometrii molekuly, spočíst její energii, vibrační frekvence, IR spektra a další, s použitím semiempirických nebo ab initio metod a s použitím nejrůznějších bázových setů. Gamess samotný nemá žádné GUI, s programem je možné komunikovat pouze přes příkazovou řádku. Existují tedy v zásadě dvě možnosti, jak mu předat vstupní data a spustit výpočet: buď manuálně vytvořit vstupní soubor[c] a nebo používat nějaký další vizualizační nástroj, ve kterém je možné nakreslit požadovanou strukturu, zadat podrobnosti k výpočtu a odeslat příkaz výpočetnímu programu. Lze použít například program Ghemical nebo Molden. Výpočetní možnosti programu Gamess mají jistá omezení (která se ovšem uživatele běžného PC nejspíše nedotknou): počet atomů počítané struktuty je omezen na 2000, počet bázových funkcí nesmí přesáhnout 8192. Program gamess je freeware, ale není volně šiřitelný. Je ho možné stáhnout v podobě archivu chráněného heslem, toto heslo i další instrukce ke stažení jsou poslány e-mailem po vyplnění krátkého formuláře[25].
Ghemical je programový balík pro kvantově chemické výpočty. Skrývá v sobě grafické rozhraní, ze kterého mohou být souštěny ab initio (zajišťované programem MPQC) i semiempirické (zajišťované programem MOPAC) kalkulace. Ghemical je také vizualizačním nástrojem pro aplikaci Gamess, to znamená, že umí vytvářet a odesílat soubory formátu INP, které Gamess používá jako vstupní data pro své výpočty. Molekulu je možné do programu buď naimportovat a nebo přímo v programu nakreslit v 3D projekci. Kreslení je snadné a intuitivní, bohužel chybí databáze templátů, například kruhů nebo častých funkčních skupin. Lze zvolit různé druhy modelů (drátěný, kalotový...), různé barvy, s molekulou libovolně pohybovat v prostoru. Pro lepší přehlednost je možné zviditelnit pouze některé části molekuly (schovat vodíkové atomy). Program umí nechat spočítat energii dané struktury, mapu elektrostatického potenciálu, tranzitní stavy, optimalizovat geometrii a další. Dále je možné měřit vzálenosti atomů v molekule, úhly, torsní úhly, nechat vynést závislost energie struktury na torsním úhlu (dvourozměrný graf, pokud se vynáší energie oproti jednomu úhlu nebo trojrozměrný, pokud se vynáší proti dvěma úhlům) nebo reakční koordinátě, spočítat MO diagram (energetické hladiny jednotlivých orbitalů a jejich obsazení). Všechny příkazy jsou v chronologickém pořadí zobrazeny ve spodní části okna, stejně jako aktuální průběh výpočtů a jednotlivé iterace. V programu může být otevřeno několik projektů najednou (projekt je obvykle jedna molekula). I pro jednu molekulu je možné zvolit několik zobrazení (tzv. camera view) a přepínat mezi nimi, například podle toho jaký typ informace je zrovna potřeba zobrazit. Aplikaci lze ovládat i příkazovou řádkou, což ovšem předpokládá znalost poměrně velkého množství příkazů. Rychlejší je v tomto případě klasické ovládání myší.
MOPlot je program pro
vizualizaci výsledků kvantových výpočtů především programu Gaussian.
Je možné ho používat i pro ostatní výpočtový software, ale výstupní
soubory musí být nejdříve převedeny do "Gaussian" formátu[26].
MOPlot tedy slouží především pro rychlé zobrazení a pohodlnou
manipulaci s vypočtenými MO orbitaly. Veškerá grafika je vektorová, to
také umožňuje nesmírně rychlý a plynulý průběh veškerého
renderování. Umí zobrazit i vibrace, pokud to předchozí výpočet
umožňuje.
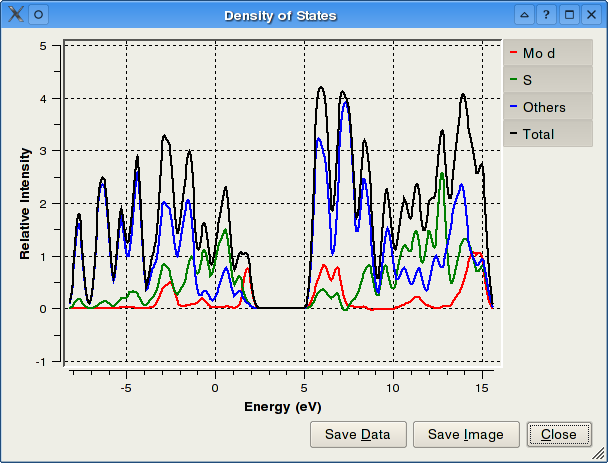
PyMOlyze je program pro analýzu výsledků DFT[d] výpočtů. Podporovány jsou výstupy programů Gaussian a Jaguar, jako vstup jsou použity soubory typu LOG, OUT, ADFOUT. Na samotný předchozí výpočet v DFT programu jsou kladeny jisté podmínky, v případě Gaussianu se musí jednat o výpočet energie (Single Point Energy) a je také požadován výpis všech informací (zadat "pop=full" v položce "Keywords"). Tento program byl inspirován softwarem AOMix (jedná se pouze o konzoli pro operační systém Windows). PyMOlyze dokáže provést následující analýzy: MPA (Mullikenova populační analýza), SCPA (C-čtvercová populační analýza), DOS (četnost jednotlivých stavů), OPA (překryvová populační analýza). Výsledkem je textový i grafický výstup.
Tinker je programový
balík, který v sobě skrývá několik desítek aplikací pro modelování a
kvantové výpočty (Alchemy, Analyze, Newton, Vibrate...). Tyto jsou pak zaštítěny grafickým
rozhraním programu Force Field
Explorer. Pomocí tohoto software je tedy možné vymodelovat požadovanou
strukturu, spočítat energii a optimalizovat geometrii, hledat tranzitní
stavy a další.
WinMopac je program pro
semiempirické výpočty. Je následovníkem staršího DOSovského programu MOPAC7[27],
obsahuje GUI, které značně
usnadní zadávání výpočtů a také integrovaný prohlížeč RasWin.
Jako vstup program požívá soubory typu MPN, tyto Z-matice mohou být buď napsány manuálně
přímo v programu a nebo může být molekula nakreslena v nějakém
externím editoru a konvertována například pomocí programu BabelWin do
formátu MPN. Stejně tak klíčová slova, tedy další podrobnosti k
výpočtu (bázové sety,
náboj struktury...) mohou být
napsána přímo do vstupního souboru nebo vybrána pomocí grafického
rozhraní. Na výběr je z několika bázových setů (AM1,
MINDO/3, PM3, MNDO...), metody výpočtu
jsou semiempirické. Výsledky se
zobrazí v úplné nebo zkrácené formě která obsahuje jen celkové výsledky,
jako spočtené spalné teplo, energii, ionizační potenciál a
několik dalších údajů. Spočtenou optimální geometrii molekuly
lze zobrazit v RasWinu a nebo načíst v externím programu.
Matematické nástroje
Extrema je multifunkční programovací rozhraní podobné například komerčnímu programu Matlab[28]. Vzhled hlavního okna je jednoduchý a přehledný, s programem se komunikuje pomocí krátkých textových příkazů. Program lze využít jako pokročilou kalkulačku k řešení algebraických rovnic, počítání derivací a integrálů, kreslení grafů jak různých matematických funkcí tak i ze zadaných dat, prokládání regresních funkcí těmito daty a fitování parametrů. Všechny proměnné jsou (podobně jako v Matlabu) automaticky považovány za vektory (resp. matice), což usnadňuje vkládání dat. Ty lze vložit jak přes příkazovou řádku (vhodné jen pro menší kvanta), tak načíst z textových souborů. Téměř všechny parametry vykreslených grafů lze nastavit podle vlastní potřeby. Extrema ovládá přes 200 různých funkcí (od logaritmů a goniometrických funkcí až po Gaussovy, Fermiho-Diracovy, error funkce, Furierovu transformaci či Hermitovy polynomy) a přes 30 operátorů. Lze požívat vlastní skripty, ukládat je pro pozdější použití nebo používat skripty už tvůrci programu přednastavené. Extrema umožňuje jednoduché programování a práci s matematickou analýzou, a to v poměrně příjemném grafickém prostředí a v jednoduchém a srozumitelném jazyce. V programu je k dispozi rozsáhlá nápověda.
FreeMat je
chemicko-inženýrský nástroj pro nejrůznější komplikované matematické
výpočty. Jedná se vpodstatě o další klon Matlabu a od svých
vrstevníků (Scilab, Octave) se liší poměrně málo. Záleží
vpodstatě jen na vkusu uživatele pro který nástroj se rozhodne. FreeMat
dle autorů v současnosti disponuje asi 95 % funkcí Matlabu, ale
stále se vyvíjí.
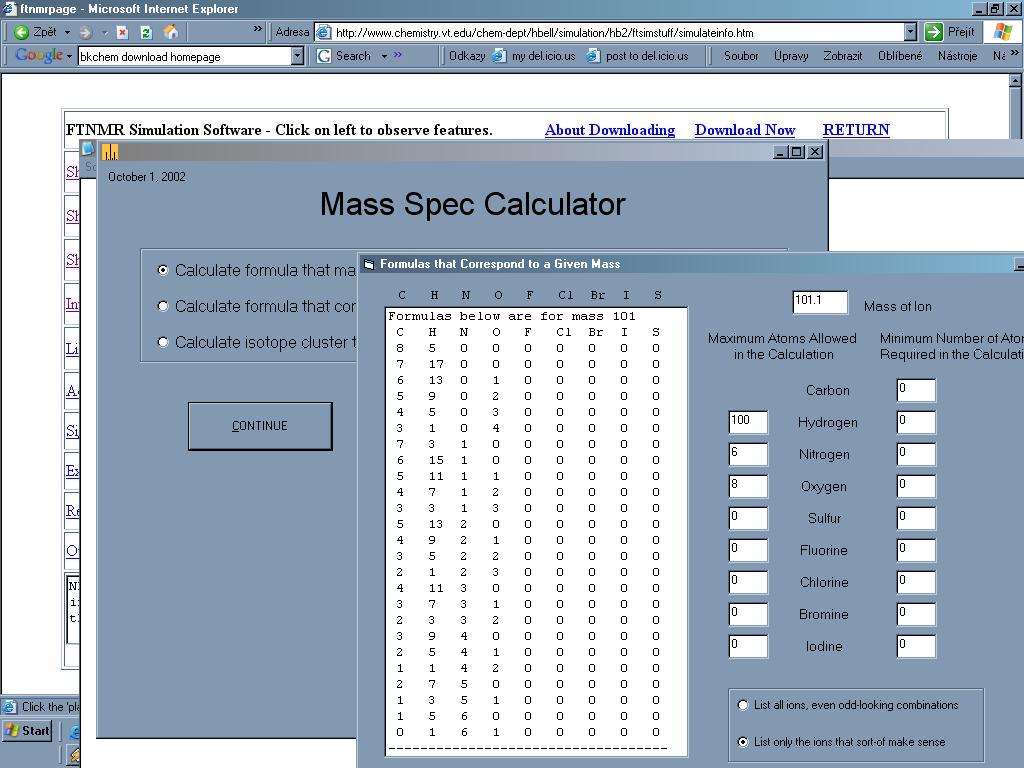
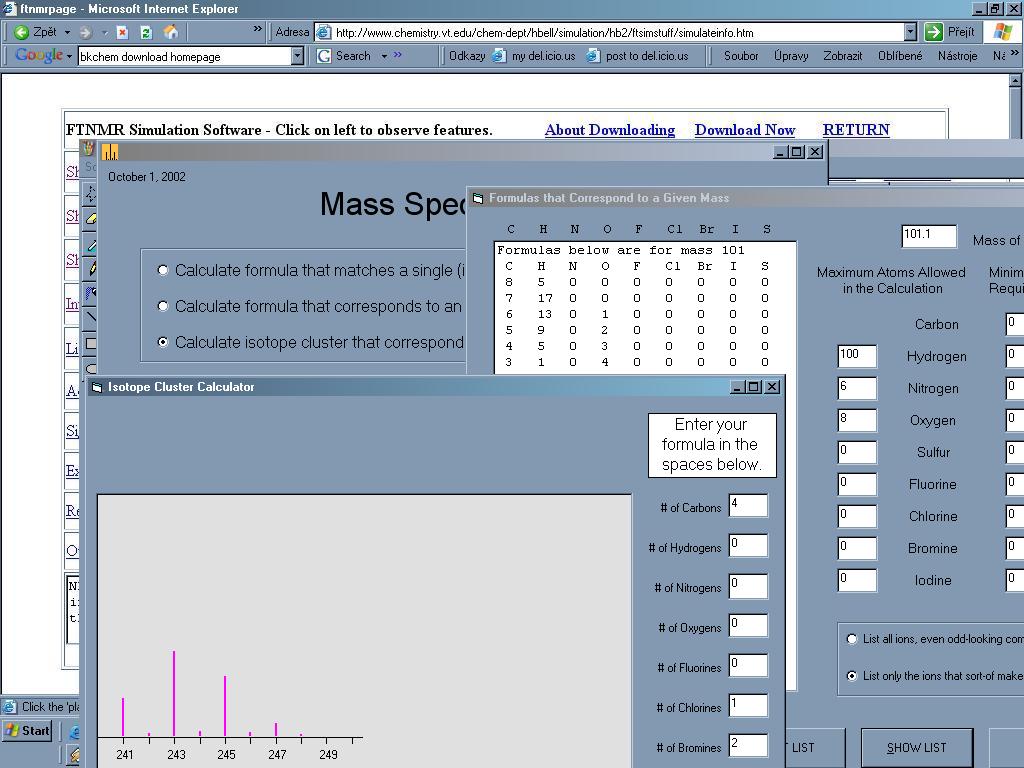
Mass Spec je
užitečný nástroj který pomůže s analýzou MS spektra[29].
Dokáže spočítat všechny možné sumární vzorce ze zadané přesné
hmotnosti iontu, pokusí se o analýzu spektra (po zadání všech hmotností a
intenzit iontů navrhne alternativy), dokáže předpovědět
vzhled izotopových píků.
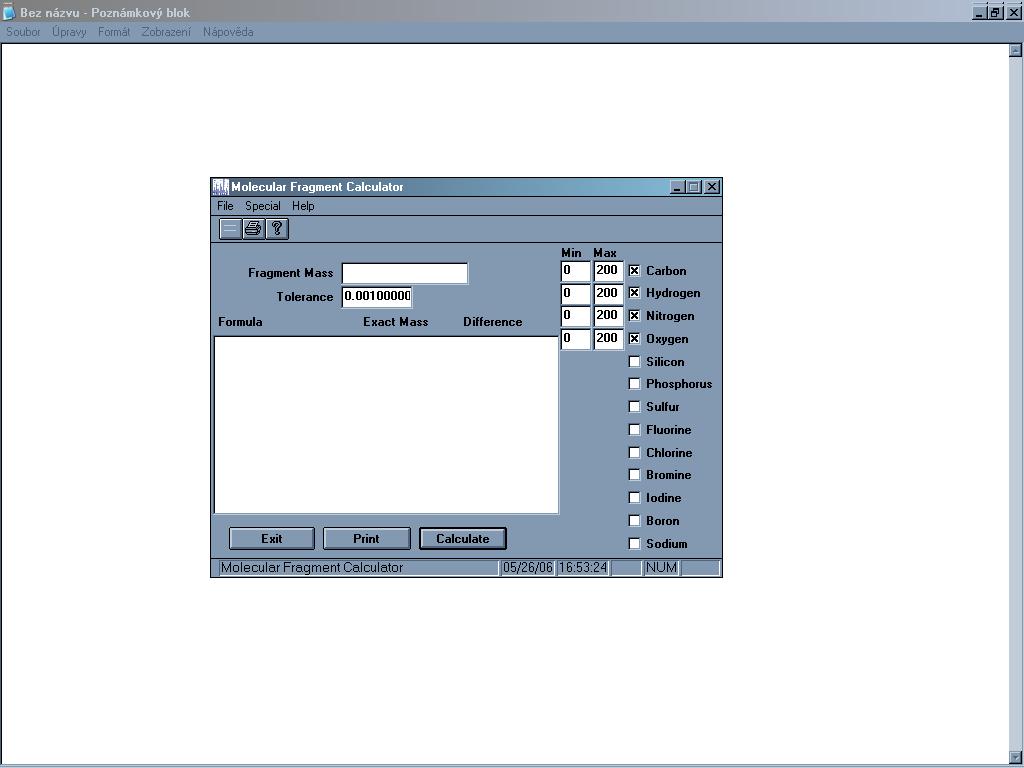
Jednoduchý program, který umí ze zadané přesné relativní molekulové
hmotnosti spočíst sumární vzorec sloučeniny, popřípadě více
vzorců (lze zvolit tolerovaný rozsah), u každého vypíše přesnou
hmotnost a odchylku od zadané. Lze vybrat, které prvky mají být ve
sloučenině zastoupeny (vybírat lze ale jen z 13 nejběžnějších).
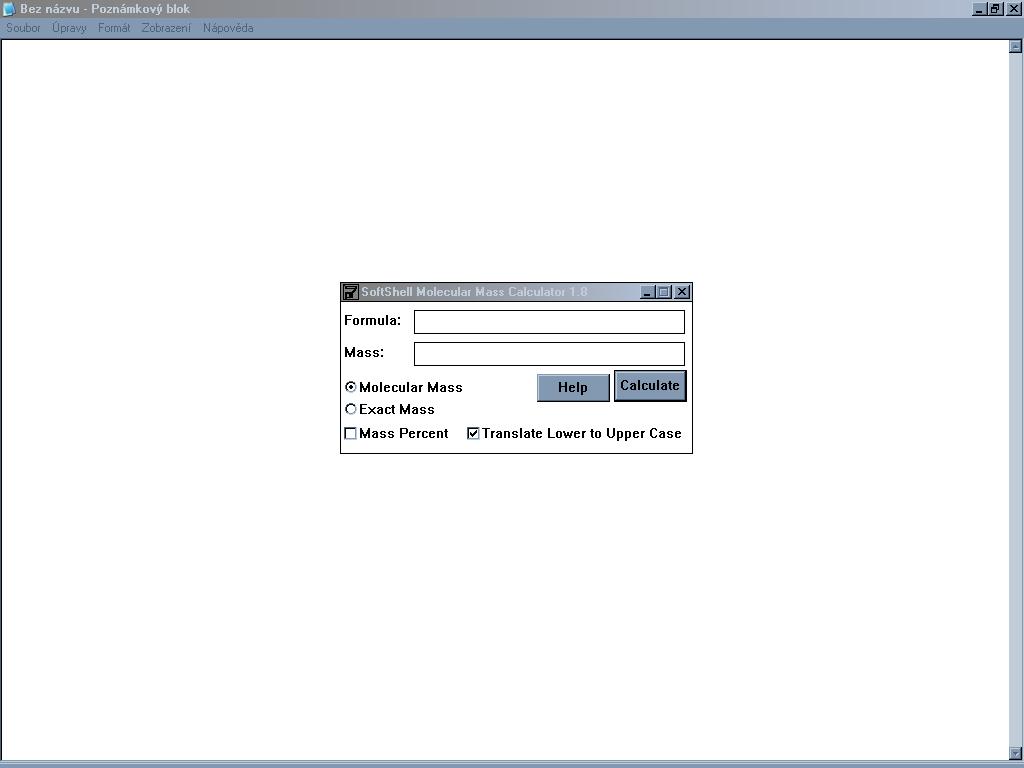
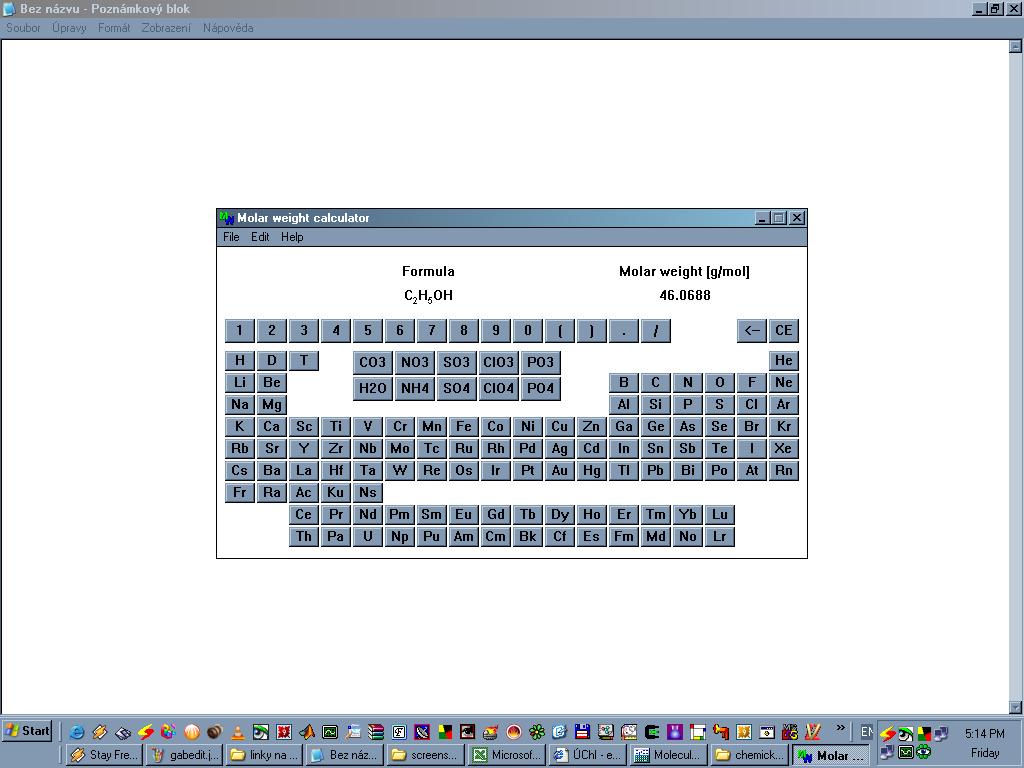
Velmi jednoduchý program, který ze zadaného sumárního vzorce spočítá
přesnou molekulovou hmostnost a procentuální zastoupení prvků.
Tato aplikace dokáže ze zadaného sumárního vzorce sloučeniny
spočítat relativní molekulovou hmostnost a procentuální zastoupení
prvků. Identifikuje i zkratky
aminokyselin či obvyklých ligandů (Gly, Ala, Bpy..)[30].
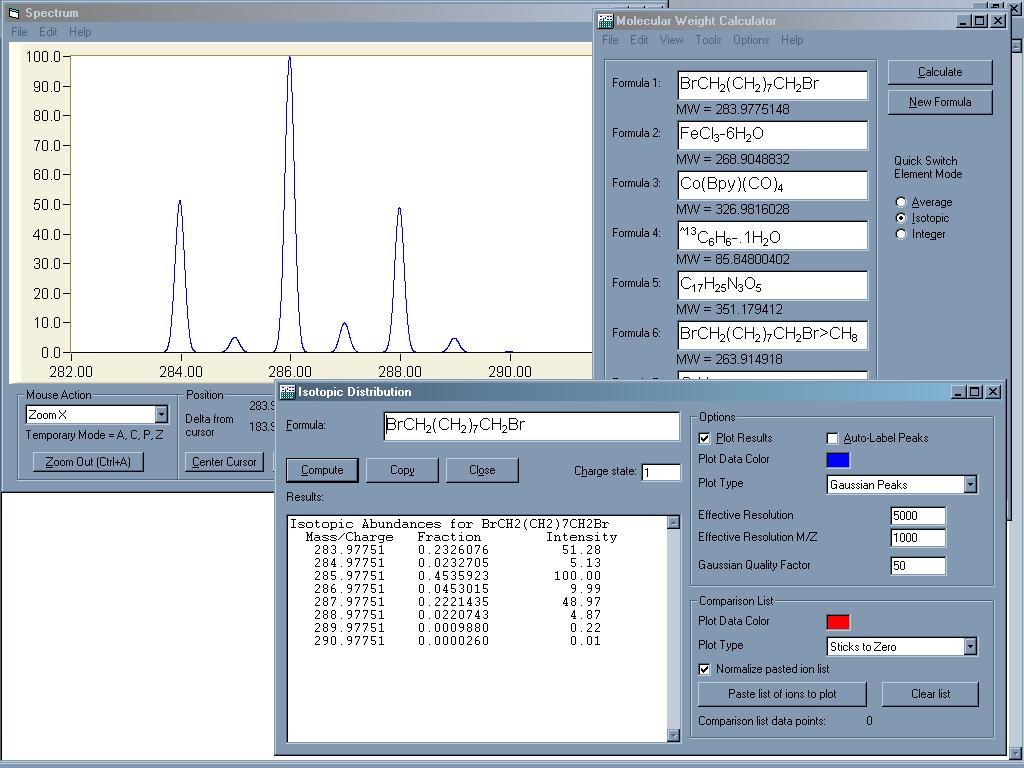
Je možno zvolit mezi různým izotopickým či průměrným
složením. Program dokáže i opak - pro zadanou relativní molekulovou hmotnost navrhne
různé varianty sumárního vzorce. Je možno zvolit jaké prvky mají být
zastoupeny a jaká je povolená odchylka. Program vyhodnotí možné relativní molekulové hmotnosti dané sloučeniny
s přihlédnutím k přírodnímu výskytu izotopů a výsledky zobrazí v
podobě Gaussovských píků závislosti pravděpodobnosti na
hmotnosti (lze užít například pro vyhodnocení některých molekulových
píků v hmotnostní spektrometrii). V programu je i jednoduhé makro pro rutinní výpočty koncentrací -
ředění roztoků, molární a hmotnostní koncentrace.
GNU Octave je
matematický nástroj, který umožní numerické řešení složitých chemických a
inženýrských problémů. Řešení lineárních i nelinárních rovnic,
diferenciálních rovnic, složitých soustav, kreslení grafů, to je jen
zlomek z jeho vlastností. Uživatel si může napsat své vlastní programy
pomocí vestavěných operátorů a funkcí. Octave a jeho jazyk je již
tradičně kompatibilní se svým vzorem, programem Matlab, lze
ale používat i moduly napsané v C++ nebo Fortranu. GUI je o něco méně
přehledné než u podobných aplikací Scilab a FreeMat. K dispozici jsou
prekompilované instalace pro Windows, Linux a Mac OS.
Scilab je další propracovaný matematický software podobný předchozímu. Dokáže spolupracovat i s dalším známou komerční aplikací Maple, obsahuje kódy Maplovských funkcí, které umožní přenést objekty z Maplu do Scilabu. Obdobně je kompatibilní i s Matlabem. Po spuštění se zobrazí konzole, ve které mohou být zadávány příkazy jako v klasické kalkulačce a spouštěny nejrůznější funkce. Ve verzi 4.0 je to celkem 1579 přednastavených funkcí. Jen ve velmi stručném výčtu: program Scilab umí například kreslit 2D a 3D grafy, vytvářet animace, ovládá algebru, matice, integrování, statistiku a má mnoho dalších funkcí. Vzhledem k tomu, že se jedná o Open Source, mohou být uživateli libolné funkce a vlastnosti dále přidávány. Asi nejzajímavější vlastností je opět možnost si v externím editoru pomocí jednoduchých příkazů napsat program, který potřebujeme na vyřešení daného problému, a ten pak spustit ve Scilabu. Přestože příkazy a syntaxe se od Matlabu natož Maplu a ostatních liší, struktura je v zásadě totožná, k dispozici je navíc obsáhlý help a množství tutorialů. Scilab vytváří zcela plnohodnotnou alternativu výše zmiňovaným komerčním programům.
Zpracování experimentálních dat
ECOMAC je program pro ovládání chomatografických zařízení, úpravu a ukládání naměřených dat. Jedná se o Open Source software vyvinutý ve firmě ECOM především pro komunikace se zařízeními její výroby, lze ovšem použít i pro přístroje některých jiných značek. Program slouží pro komunikaci s chromatografickými zařízeními (pumpy, detektory, temperační zařízení, teploměry) a pro manipulaci s naměřenými daty.
Fityk je program pro prokládání série experimentálních bodů regresními funkcemi. Data mohou být proložena obecně nelineární funkcí (Gaussova křivka, Lorentzova funkce, polynomy...) a jsou fitovány (zjišťovány jejich parametry). Program je obzvláště vhodný pro funkce tvaru jednoduchých píků, jejich plochy lze automaticky integrovat, nalézt maximum, výšku, pološířku pásu. Dle autorů nachází program uplatnění zejména v oblastech krystalografie, chromatografie, a infračervené a Ramanovy spektroskopie. Dá se použít i prostému zobrazení a prohlížení naměřených dat. S body je možno libovolně manipulovat, zanedbávat odlehlé hodnoty, zobrazit jen potřebné části spekra. Program má jednoduché a snadno ovladatelné grafické rozhraní, krom toho disponuje i konzolí, kde je možno provádět všechny uvedené a další operace pomocí jednoduchých příkazů.
FTNMR, jednoduchý program napsaný ve Visual Basicu, umí načíst a zpracovat FID z NMR experimentu. Jsou podporovány tyto typy dat: 1. JCAMP spektrální data 2. JCAMP FIDy 3. NUTS FIDy, Typy 1, 2, & 3 4. Varian Unity FIDy 5. Anasazi (LYBRICS) FIDy 6. FIDy uložené jako textové soubory Program provede Fourierovu transformaci, zfázuje spektrum, umožní ho dále upravovat, uložit a vytisknout.
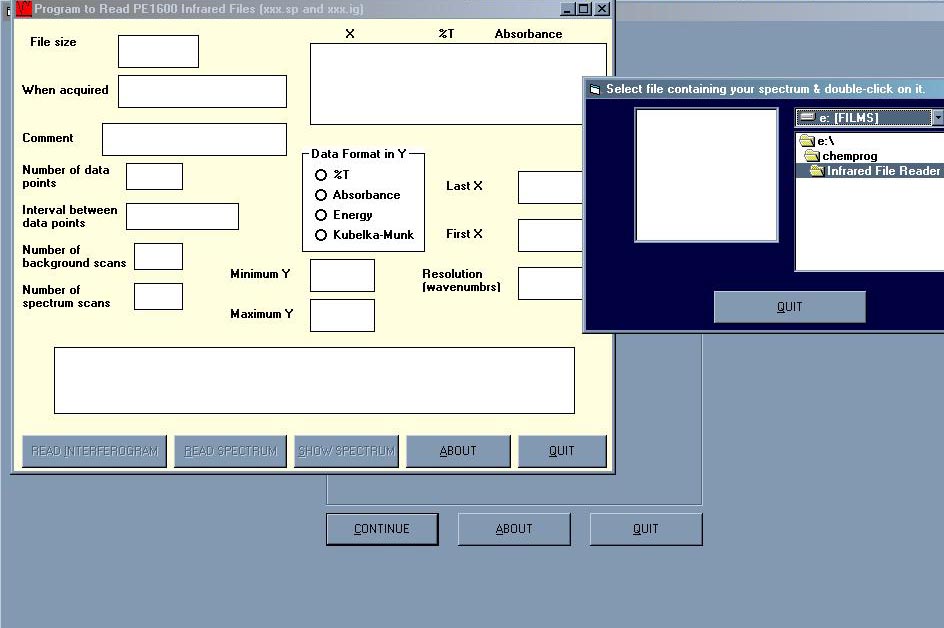
Infrared File Reader umí načíst a zpracovat IR data těchto formátů: SP, IG, JDX, SPC, SPA, TXT (comma-separated X,Y points). Spektrul lze zpracovat, zobrazit několik látek na sebe pro porovnání, uložit a vytisknout.
Program pro zpracování a analýzu experimentálních dat. Data lza načíst z různých souborů textového typu, program podporuje například i excelovské CSV soubory, pro vložení dat lze použít i prostou metodu copy-paste. Vložená data lze vykreslit do grafu, ten upravit podle potřeby a data mnoha způsoby dále zpracovávat. Mezi nabízenými možnostmi je například proložení regresní křivkou, derivace, filtrace šumu, korekce základní linie, klastrová analýza. Pro zpracování velkého objemu dat (řádově desítky tisíc) lze použít redukující funkci PCA (Principal component analysis) která usnadní a urychlí další zpracování. K dispozici jsou další statistické funkce jako například DFA (Discriminant function analysis), GA-DFA (A genetic algorithm coupled to discriminant function analysis), GA-PLSC (A genetic algorithm coupled to partial lest square calibration). Data lze v kterémkoliv okamžiku uložit ve speciálním PyChem formátu a v přerušené práci kdykoliv pokračovat.
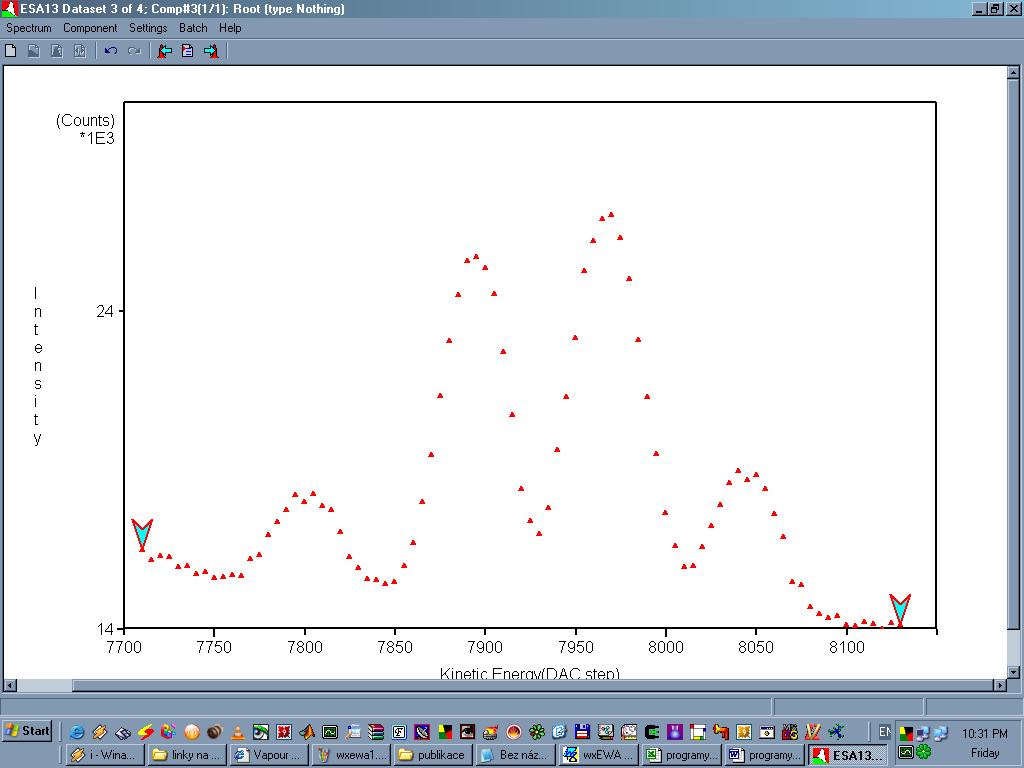
wxEWA je nástroj pro vyhodnocování spektroskopických dat. Program tato data dokáže importovat v podobě souborů ve formátu DAT. Spektrum se automaticky zobrazí ve zvláštním okně v podobě jednotlivých experimentálních bodů. Experimentální body lze proložit křivkou a fitovat parametry, je zahrnuto i statistické zpracování a výpočet nejistot. Při pohybu kursoru v tomto okně se automaticky zobrazují souřadnice (to se hodí například při odečítání hodnot). Libovolnou část grafu lze jednoduše označit a zvětšit. Program má mnoho funkcí, které usnadní interpretaci spektra. Samozřejmostí je analýza píků (polohy, maxima, pološířky, plochy), dále lze například nezávisle spočíst pološířky a plochy dvou píků i pokud se tyto částečně překrývají. Osy grafů se dají zobrazit v logaritmických (semilogaritmických) souřadnicích. Pomocí funkcí Undo a Redo je možné snadno odvolat všechny nechtěné operace a úpravy. Všechny tyto operace a úkoly se přehledně zobrazují do zvláštního okna (log), ze kterého je jasné kdy a v jakém pořadí byly dané příkazy provedeny. Upravené spektrum (i všechny výsledky a log) je možné uložit i vytisknout.
Ostatní
BabelWin je GUI pro známý program Babel. Pro jeho chod je tedy mít třeba nainstalované programy Babel 1.06 a Babel 1.6. Vhodně se doplňuje s programem OpenBabel, zvládá některé formáty, které nejsou v OpenBabelu obsaženy. BabelWin umí vytvořit soubory těchto typů: Alchemy Ball and Stick Cacao Cartesian CAChe MolStruct* Chem3D Cartesian 1 Chem3D Cartesian 2 ChemDraw Conn. Table* CSD CSSR* Gamess Input* Gaussian Cartesian* Gaussian Z-matrix Hyperchem HIN* IDATM Mac Molecule Macromodel Micro World MM2 Input* MM2 Ouput MM3* MMADS* MDL Molfile* Mopac Cartesian Mopac Internal PDB Report Sybyl Mol* Sybyl Mol2* XYZ
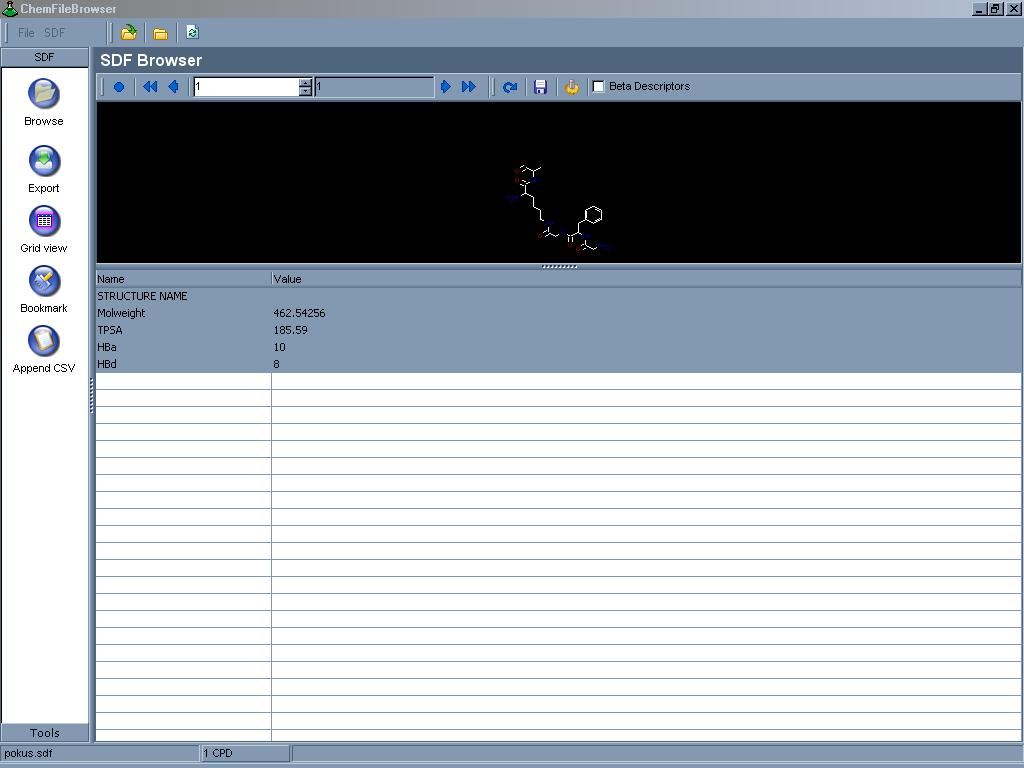
Prohlížeč pro práci se SDF soubory. Usnadní práci s velkým množstvím
dat uložených právě v tomto formátu, umožní jednoduché prohlížení a export
do jiných aplikací, zobrazuje současně některé informace o
molekule, jako relativní molekulovou hmotnost a HbD, HbA, TPSA deskriptory.
ChemFormatter je zásuvný modul k programovému balíku Microsoft Office, který usnadní psaní chemických vzorců a formátovaní chemických dokumentů. Po nainstalování se v programech Word, Excel a PowerPoint objeví programová lišta se speciálními tlačítky. Tedy, například text H2SO4
lze jedním kliknutím změnit na H2SO4, K+Cl- na K+Cl-
a podobně. Umí automaticky naformátovat názvy organických sloučenin a
reference, tedy například: "J. Am. Chem. Soc. 2003, 123(45),
6789." převede na správný "J.
Am. Chem. Soc. 2003, 123(45), 6789."; " J. Am. Chem. Soc., 123, 4567
(1985)." na správný "J.
Am. Chem. Soc., 123, 4567 (1985)." a podobně. Program neumí nic, co by nešlo v daném editoru udělat "manuálně", jen práci urychlí a usnadní.
Chemická kalkulačka pro počítání gravimetrických faktorů,
ředění a podobně. Výpočty jsou elementární, ale program
může značně urychlit rutinní práci. K dispozici je zatím v
angličtině, němčině a francouzštině.
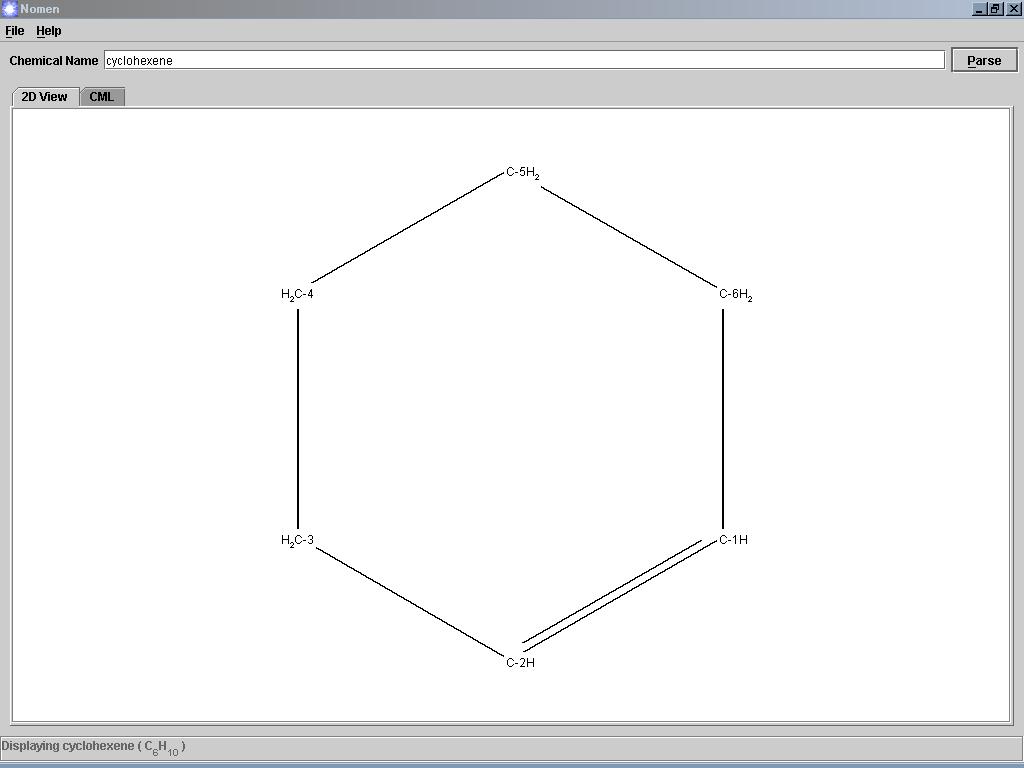
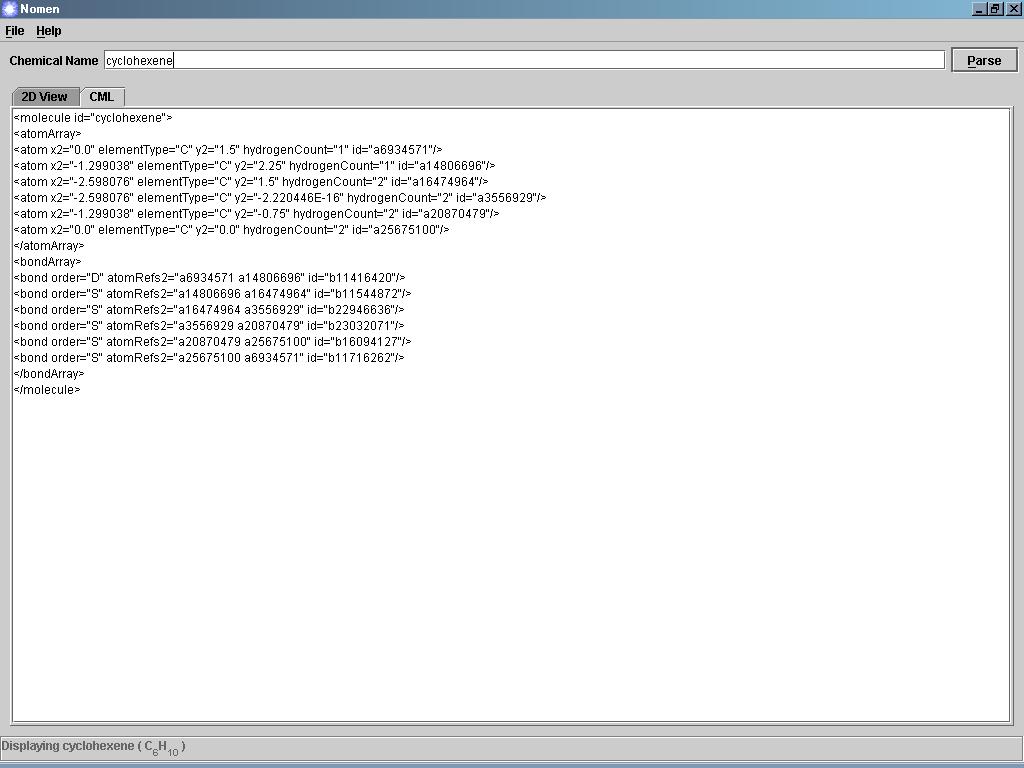
Nomen je jednoduchý program, který dokáže nakreslit chemickou sloučeninu podle zadaného systematického názvu (podle standartu IUPAC, triviální názvy nejsou bohužel podporovány). Názvy se zadávají v angličtině. Program nakreslí 2D strukturní vzorec a také popíše sloučeninu v jazyce CML. Jako soubor ve formátu CML lze danou látku i uložit. Program potřebuje nainstalované rozhraní JRE.
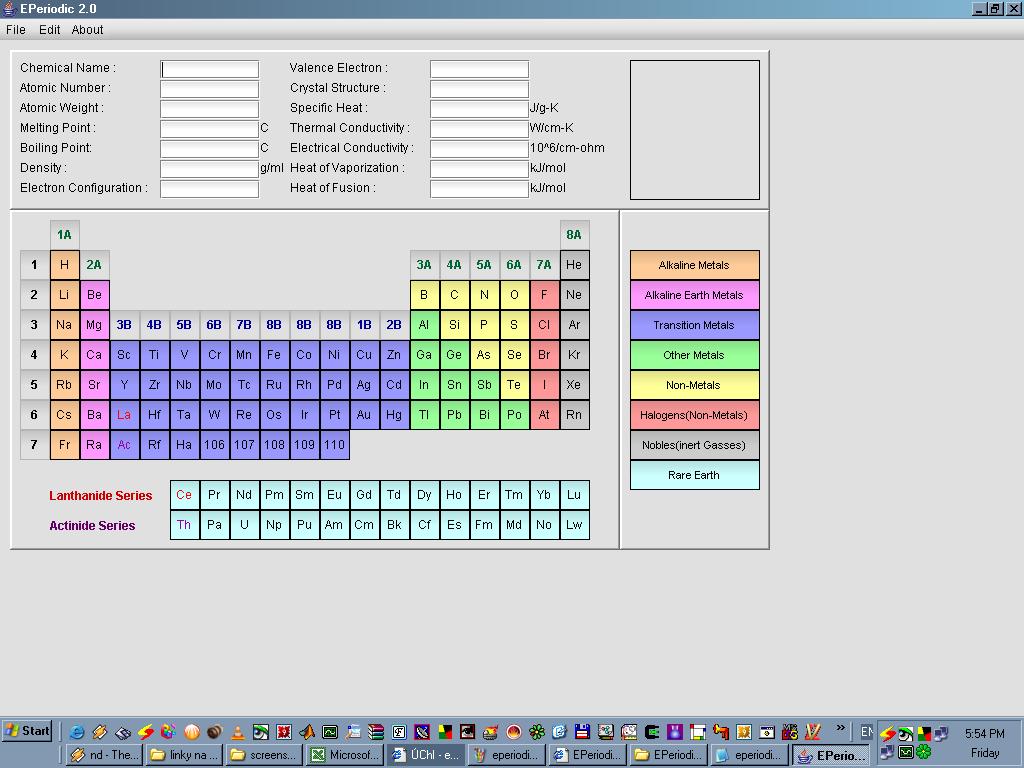
Periodická tabulka s informacemi o relativních atomových hmotnostech, bodech tání a varu, hustotách, elektronových konfiguracích, krystalových strukturách (pokud je prvek pevná látka) slučovacích teplech, tepelných a elektrických vodivostech, výparných molárních teplech a molárních teplech tání. Informace lze exportovat do XML.
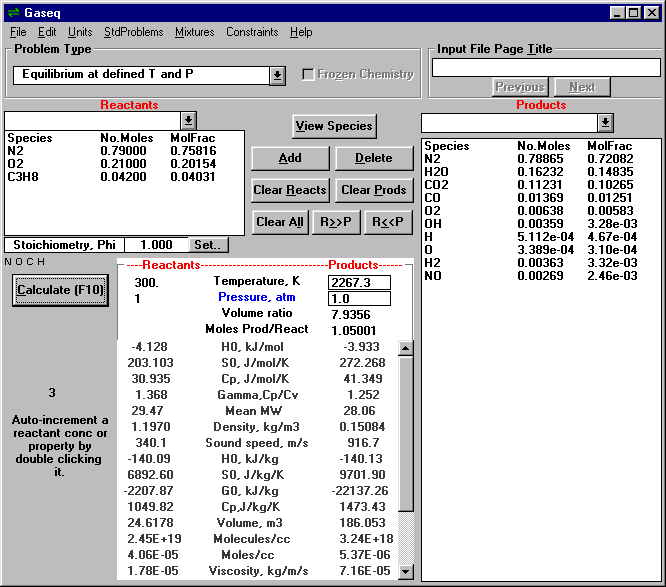
Program vhodný pro některé chemicko-inženýrské výpočty týkající
se hoření plynů a směsí plynů. Umí ze zadaných podmínek
spočítat například teplotu plamene, optimální složení směsi,
rychlostní konstanty reakcí, viskozity plynných směsí a další.
Tento program slouží k prohlížení a úpravám obrázků nejrůznějších bitmapových formátů. Je možné je libovolně editovat, měnit jejich formát, aplikovat na ně nejrůznejší filtry, vytvářet animované gify ze sekvencí. Program neobsahuje tolik funkcí jako například Paint Shop Pro nebo jiné propracované editory, ale pro základní operace s grafikou postačí.
Slouží k otevření, editaci, analýze a ukládání obrázků různých formátů. Umí dále například spočítat obsah označené plochy či velikost úhlů, dovede základní operace s barvami. Obsahuje vlastní skriptovací jazyk, který náročnějšímu uživateli umožní například nadefinovat si vlastní úpravy či zautomatizovat po sobě jdoucí série úprav obrázků.
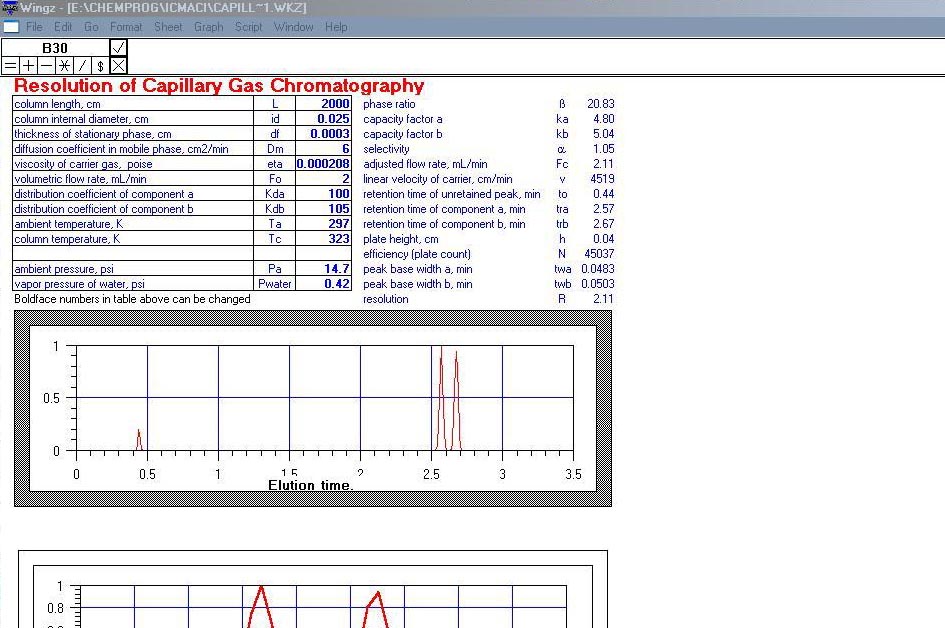
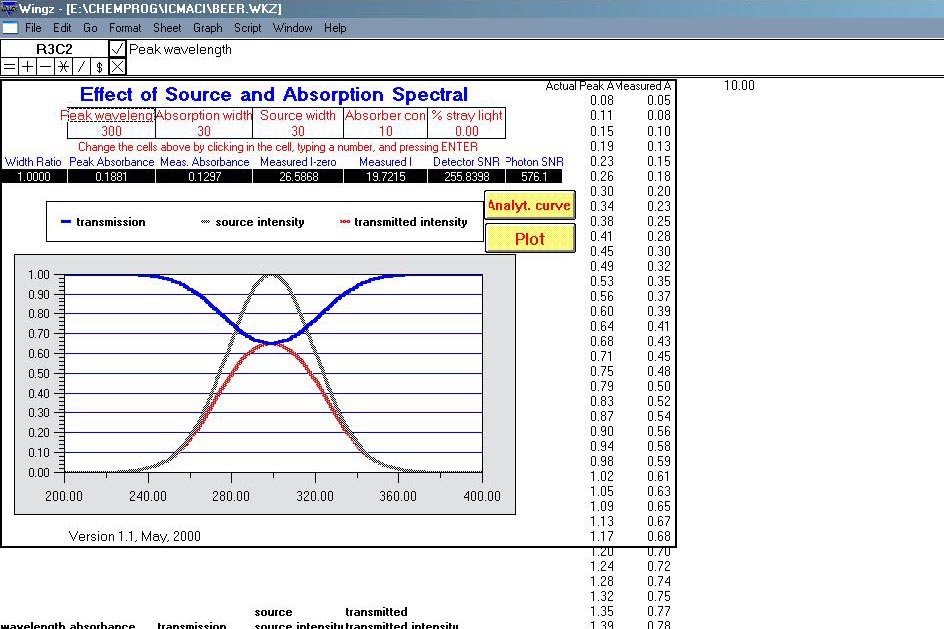
ICM je soubor modelů a simulací z prostředí instrumentální analytické chemie. Jedná se o data ve formátu WKZ, která mohou být spuštěna například v programu Wingz. Tyto interaktivní aplikace mají pouze didaktickou hodnotu, umožní se "nanečisto" seznámit s některými měřeními jako AAS, fluorescenční spektroskopie, UV/VIS spektroskopie, plynová chromatografie chromatografie a mnoha dalšími.
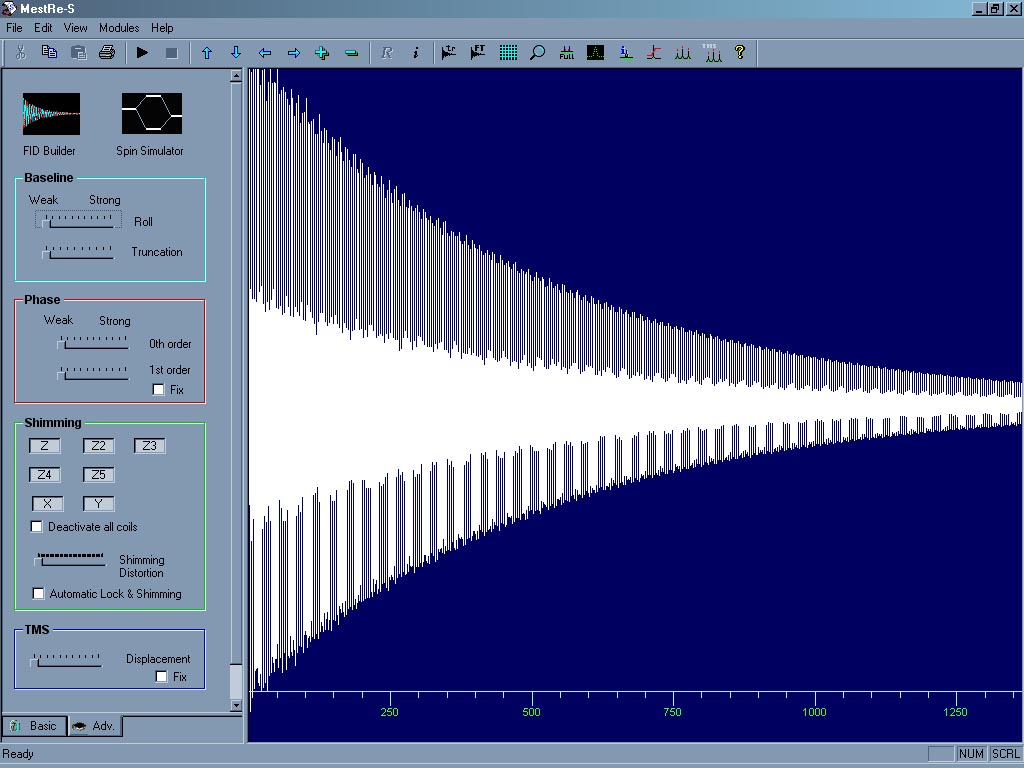
Simulátor NMR spektrometru (typu Varian). Přestože je mnoho programových rozhraní pro ovládání NMR spektrometrů a většina z nich pracuje v systému Linux či UNIX, tento jednoduchý program alespoň začátečníku umožní udělat si základní představu o postupu při získávání NMR spekter. Program je freeware, ale pro stažení je zapotřebí se zaregistrovat[31].
Interaktivní periodická tabulka. Klikáním na jednotlivé prvky nebo psaním
na klávesnici se vytvoří vzorec sloučeniny a zobrazí jeho přesná
relativní molekulová hmotnost.
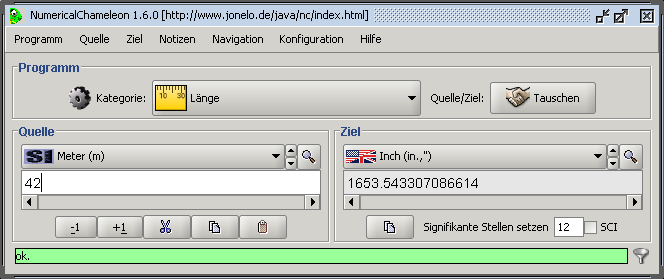
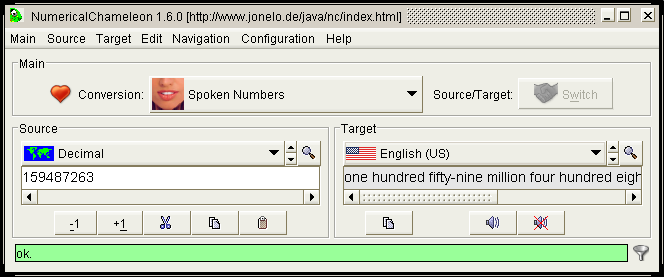
Numerical Chameleon je užitečný program pro převody jednotek. Momentálně podporuje 3276 různých jednotek 82 nejrůznějších kategorí nejen z oblasti chemie a fyziky. Lze převádět formáty data, měny, rozlišení, času, jednotky délek a ploch, teploty, síly, elektrického proudu, elektrického odporu, elektrostatického potenciálu, kapacitance, vodivosti, energie, intenzity záření, síly, hmotnosti, hustoty, magnetické indukce, radioaktivity, dynamické i kinematické viskozity, objemu, tvrdosti vody, úhlů a mnoha dalších. Seznam se bude postupem času pravděpodobně rozšiřovat, další kategorie lze stáhnout z webových stránek programu. Ovládání je velice jednoduché, z rolovacího menu stačí vybrat příslušnou kategorii a jednotku a napsat hodnotu, převedená hodnota se automaticky spočte. Všechny údaje lze průběžně ukládat do přehledného seznamu který lze libolně editovat (včetně použití HTML tagů) a uložit ve formátu TXT nebo HTML nebo data přímo přenést pomocí funkce copy-paste. Často používané převody si lze zařadit do zvláštní položky pro urychlení práce. Užitečná je funkce "Find", pokud je známa požadovaná jednotka a není jisté, zda se v dané kategorii nachází. Vzhled programu lze upravit podle mnoha přednastavených tzv. témat, komunikovat je možné celkem v osmi jazycích, čeština zahrnuta není. K programu je možné ještě zdarma stáhnout zvukové rozhraní a program BigAl, který umožní počítat požadované převody na mnohem větší počet desetinných míst. Jedná se o Java aplikace[e].
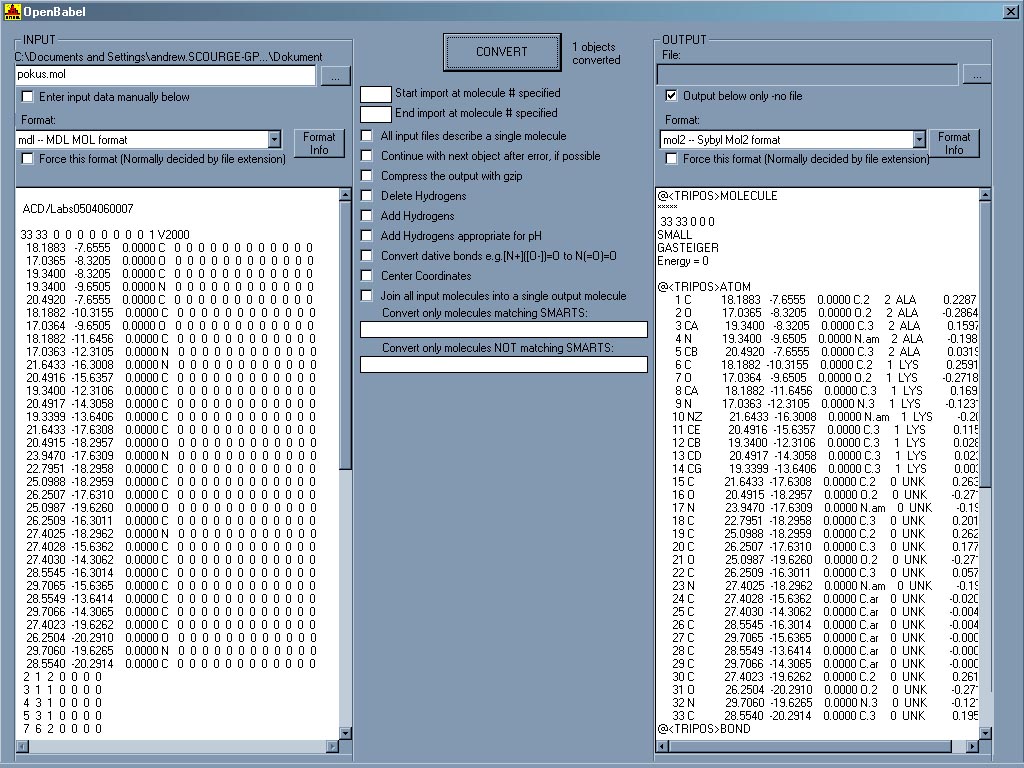
Program, který umí převádět formáty souborů používaných v chemickém modelování, výpočetní chemii a dalších oblastech mezi sebou. Podporuje většinu běžně používaných formátů. Práce se samotným programem je velice jenoduchá, stačí načíst zdrojový soubor a vybrat formát, do kterého ho chceme konvertovat. Obsah souboru se také zobrazí v textovém okně, ve kterém je ho možné editovat manuálně. Velice užitečná aplikace. Seznam podporovaných formátů:
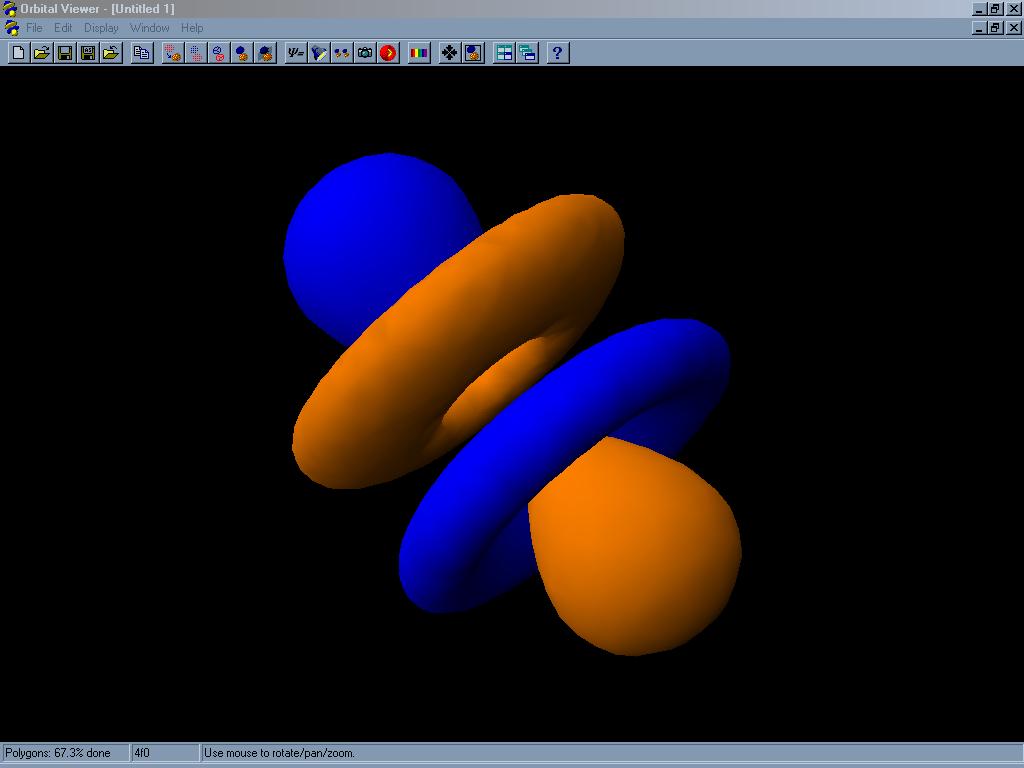
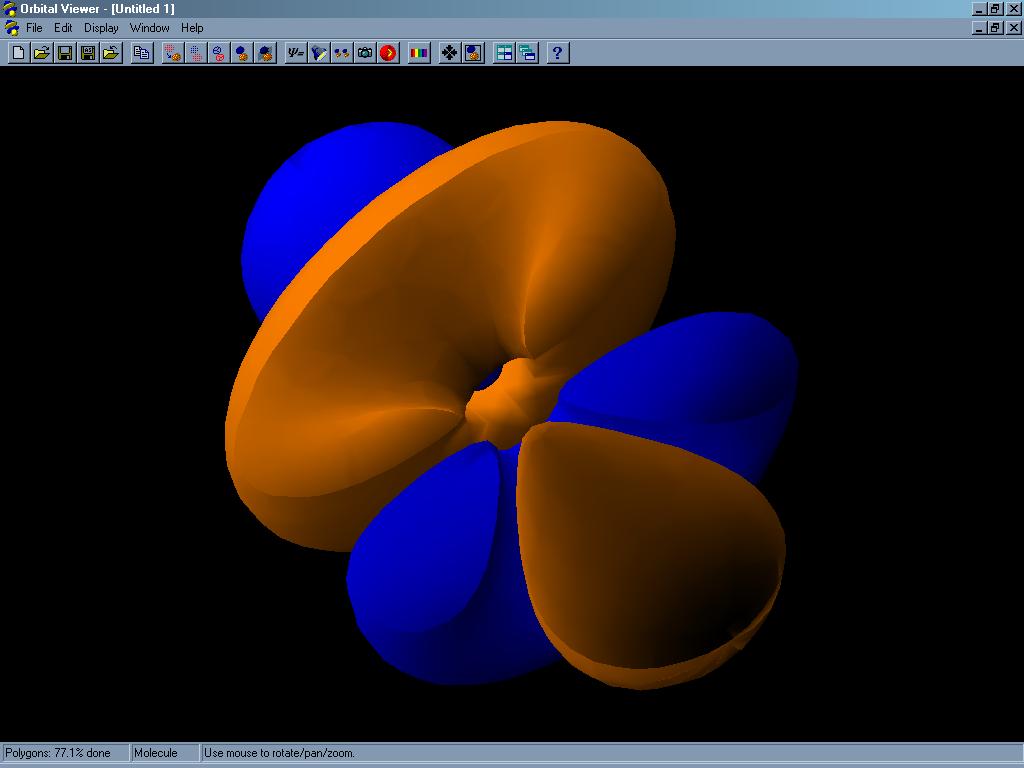
Orbital Viewer je program pro tvorbu (kreslení) atomových a molekulových orbitalů. Orbitaly jsou vlnové funkce vyhovující Schrödingerově rovnici, druhá mocnina těchto funkcí je úměrná pravděpodobnosti výskytu elektronu v okolí atomu. Program umí zobrazit orbital několika způsoby: buď jako hustotu pravděpodobnosti výskytu elektronu (čím intenzivnější barva, tím větší pravděpodobnost), jako plochu o konstantní hustotě pravděpodobnosti a nebo pomocí asymptotických ploch (ploch, na kterých je pravděpodobnost výskytu elektronu nulová). Pro větší názornost lze plochy zobrazit jako drátěný model (polygon surface) či pomocí bodů. Program umí vykreslit orbital o libovolném zadaném kvantovém čísle (přesněji o hlavním kvantovém číslo od 1 do 30 a vedlejším od 0 do 29, nad tyto hodnoty se již čas renderování stává neúnosným). Elektronová hustota v molekulách je počítána jednoduchou metodou LCAO, která počítá hodnoty vlnové funkce pro každý atom zvlášť. Spočítané orbitaly můžeme zobrazit výše popsanými způsoby, obarvit je či osvětlit z několika různých stran pro zvýraznění tvarů. Zajímavou možností je generace takzvaných stereoskopických obrazů. Program vykreslí na monitor dva obrazy téhož orbitalu z různé perspektivy, které při pohledu přes 3D brýle (stereoskop) vyvolají zdání skutečného trojrozměrného obrazu. Velmi zajímavou funkcí je možnost provádět orbitalem různé řezy, které umožní nahlédnout i do vnitřní struktury. Lze zvolit počet bodů a vlastnosti zobrazovaného povrchu a tedy vhodný kompromis mezi přesností a rychlostí výpočtu i plynulostí manipulace s vykreslenou strukturou, to ocení obzvláště uživatelé počítače s pomalejším procesorem. Pro bezprostřední porovnání tvarů několika orbitalů lze tyto seřadit za sebou do sekvence a proměnu jednoho orbitalu v druhý přehrát jako video, animace lze uložit jako AVI soubor. Orbital Viewer umí také načíst data ze souborů typu ORB (Orbital Specification Files) a OV (Orbital Viewer Files). Jedná se o textové soubory, formát OV navíc obsahuje ještě další informace o samotném renderování, což umožňuje uživateli kdykoliv pokračovat v započatém a uloženém výpočtu. Všechny vygenerované struktury je možno dále uložit ve formátech PPM, TIF, BMP, WRL nebo TXT. Posledně jmenovaný textový soubor obsahuje pouze souřadnice všech bodů vykreslené struktury v trojrozměrném prostoru.
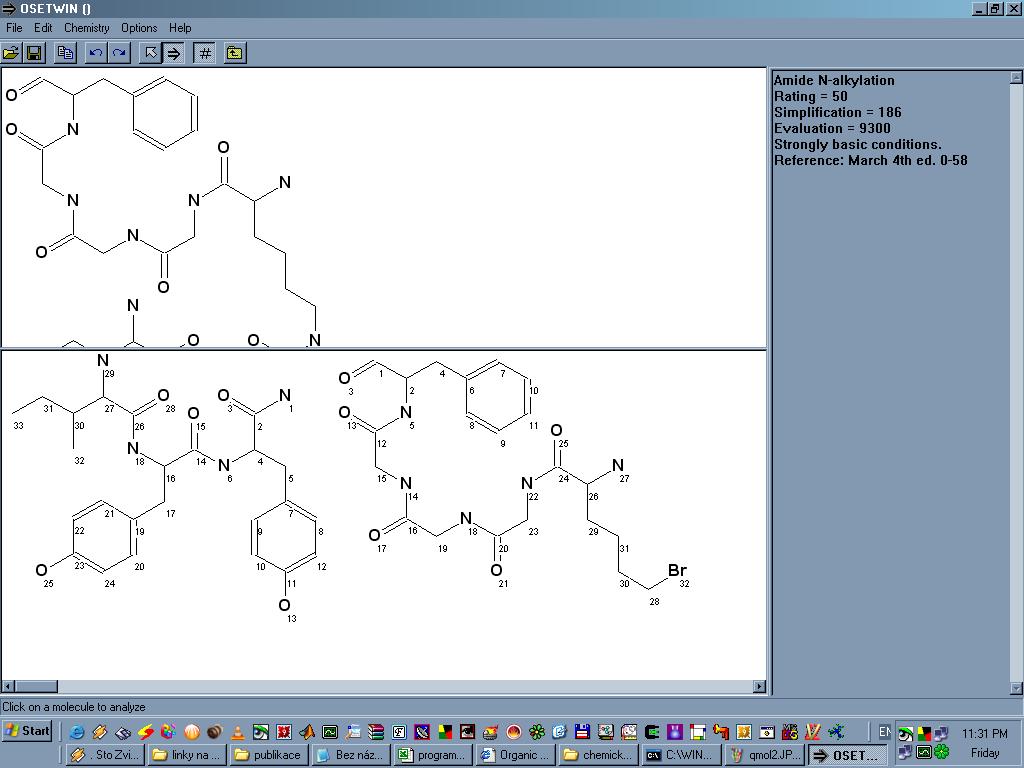
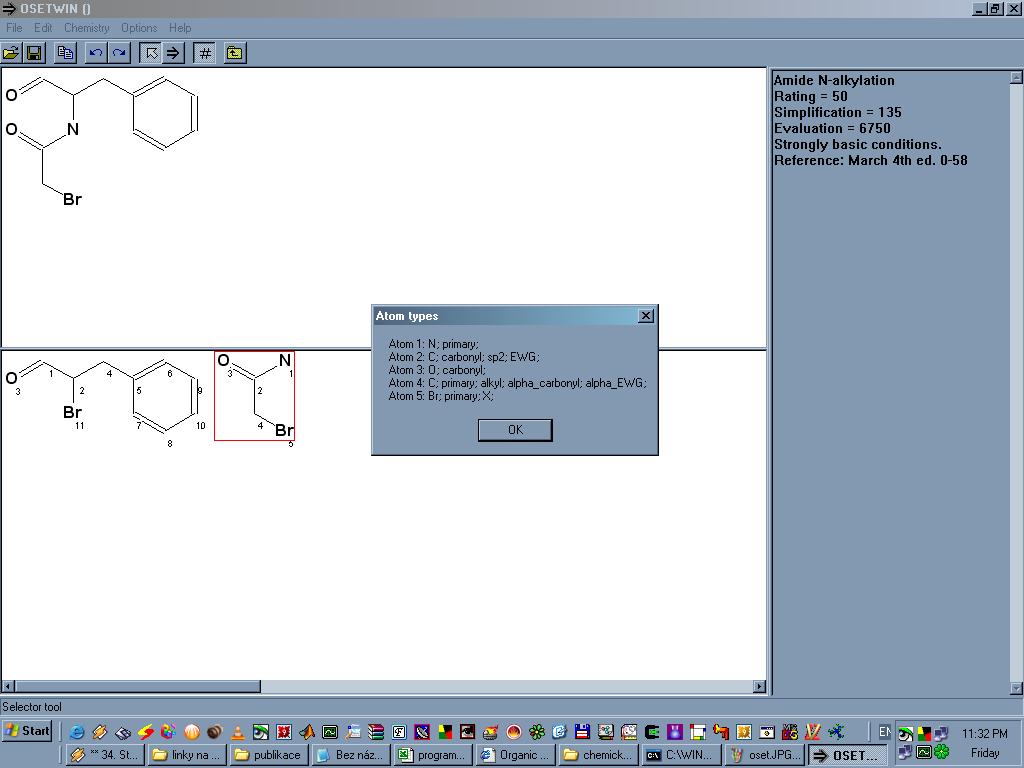
OSET je program který pomáhá v retrosyntetické analýze, tedy v myšlenkovém postupu od požadovaného produktu až k dostupným reaktantům. Produkt je nejdříve nutné nakreslit v nějakém externím editoru a uložit ve formátu MOL, ten si pak OSET dokáže naimportovat. Kliknutím na molekulu dané sloučeniny vždy dostaneme výchozí látky (látku) které OSETu připadají jako nejvhodnější reaktanty spolu s názvem dané reakce, obvyklým výtěžkem, reakčními podmínkami popřípadě dalšími poznámkami. Vytváří se tak jakýsi pomyslný "strom" na jehož vrcholu je požadovaný produkt a vespod všechny potřebné výchozí látky. U každé molekuly program umí mimojiné vygenerovat SMILES, najít ekvivalentní atomy a spočítat takzvanou "složitost" (complexity), je jasné, že směrem od produktu k výchozím látkám tato veličina klesá.
Zajímavá interaktivní periodická tabulka. Z obsažených informací lze
jmenovat například: bod varu a tání, hustotu, elektronovou konfiguraci, acidobazický charakter, krystalové
modifikace,elektronegativitu, výparné teplo a teplo tání, elektrickou a
tepelnou vodivost, měrnou tepelnou kapacitu, ionizační potenciál,
atomový poloměr. Prvky lze i hledat pomocí intervalů hodnot vybraných
(výše jmenovaných) veličin, dále lze zobrazit grafy
závislostí všech popsaných veličin na atomovém čísle.
PolyJen je simulátor polymerizačních reakcí. Na základě zadaných informací o iniciátoru a monomeru předpovídá chování reakční směsi v závislosti na čase. Program je Java aplikace, potřebuje JRE.
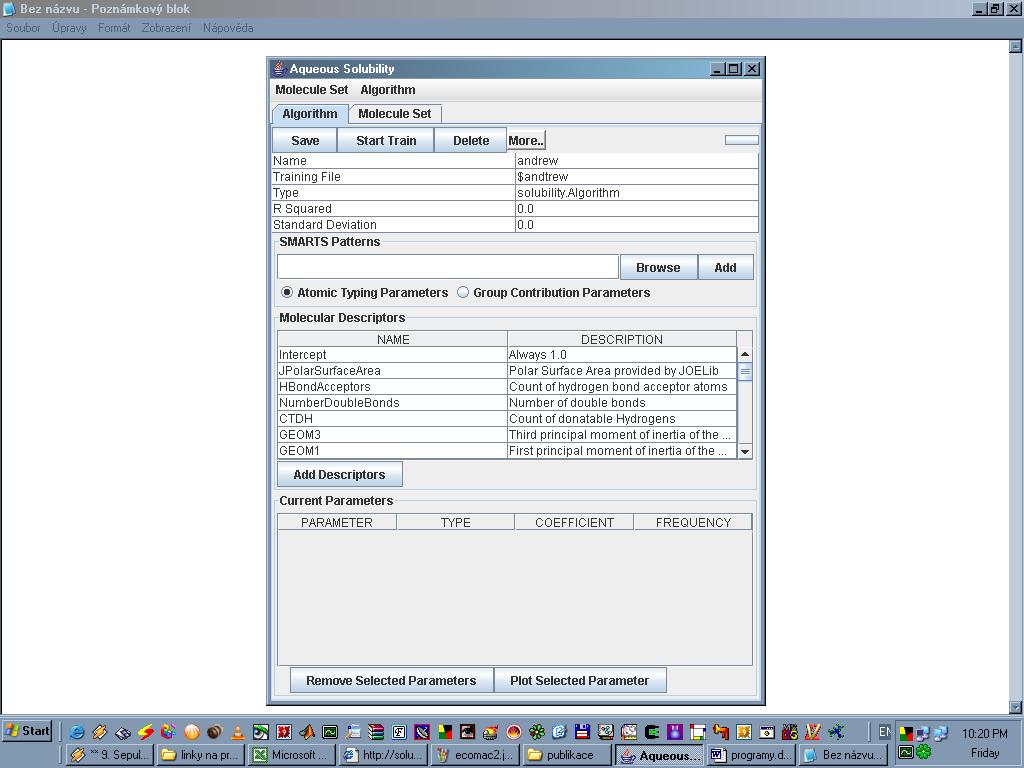
Solubility je Java aplikace sloužící k předpovídání rozpustnosti organických sloučenin v čisté vodě. Jako vstup slouží soubory ve formátu MOL2 nebo SDF. Pokud není k dispozici žádný kreslicí program, který by tyto dokázal exportovat, lze navrženou molekulu uložit v některém z běžných programů (ChemSketch, Isis Draw, WinDrawChem) a soubor poté přetransformovat na MOL2 nebo SDF například programem OpenBabel. Po importování sloučeniny (lze vložit i několik látek najednou, třeba v případě, kdy je vhodné vidět výsledky vedle sebe pro porovnání), zvolí se algoritmus (z několika přednastavených) a spustí výpočet. Program Solubility ke svému chodu potřebuje nainstalovaný JRE a to alespoň verze 1.4.
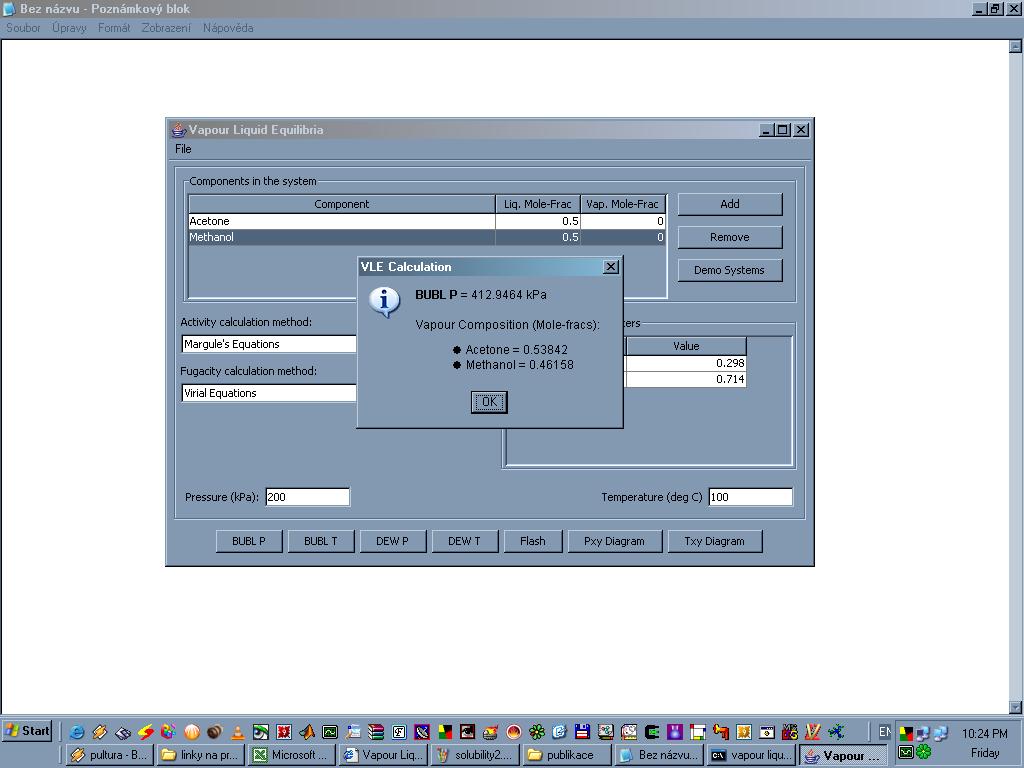
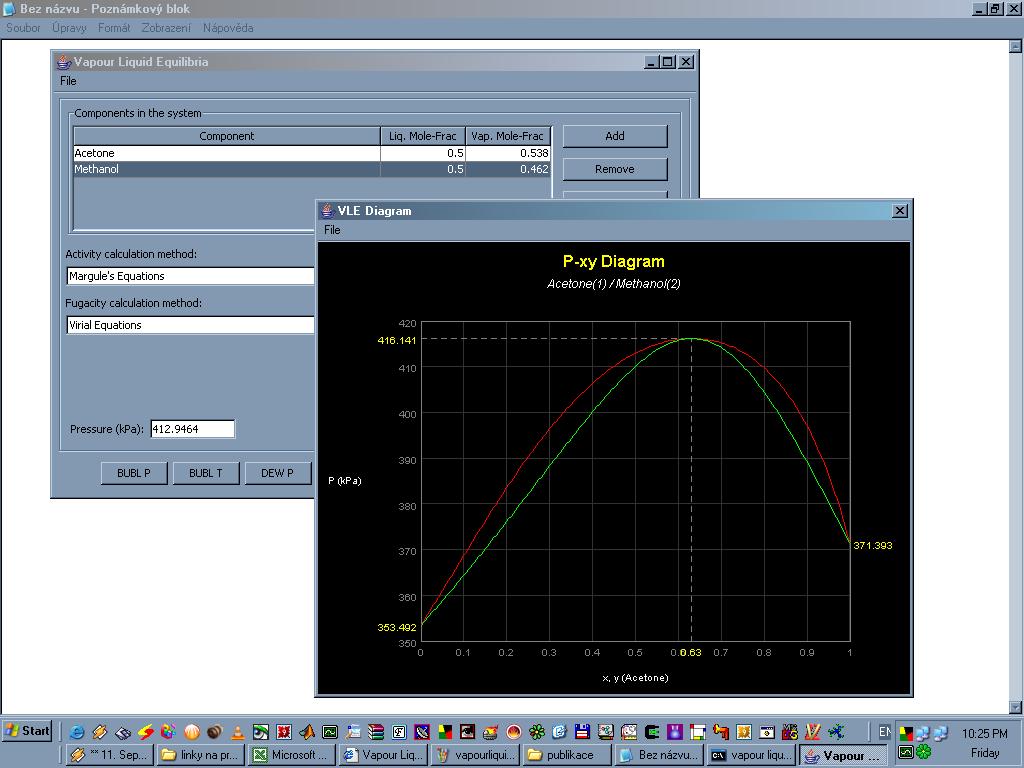
Vapour Liquid Equilibria je program napsaný v jazyce Java a ke svému chodu potřebuje nainstalované rozhraní JRE. VLE simuluje binární systémy kapalina-pára a slouží k výpočtu některých termodynamických dat. K výpočtu jsou používany empirické rovnice (Margulova, Van Laarova, Redlichova-Kwongova a viriální rovnice). Kombinaci těchto rovnic lze zvolit a porovnat dosažené výsledky. Program se používá ve výpočtu vlastností některých binárních směsí (nebo i čistých látek), převážně organické povahy. Při zadaném tlaku lze spočítat bod varu a rosný bod a naopak při zadané teplotě tlaky v těchto dvou bodech. Lze také vykreslit grafy p=p(x,y) a T=T(x,y). K dispozici je asi třicet kombinací binárních směsí (aceton-methanol, aceton-benzen, ethanol-kyselina octová, methanol-hexan... a další). Lze vytvářet i vlastní směsi, potom je ovšem nutno vložit empirické koeficienty do příslušných termodynamických rovnic (tyto koeficienty je třeba nalézt jinde, v literatuře nebo na webu). Nelze počítat vlastnosti směsí obsahující více než dvě látky.
WINDNMR je program pro simulaci NMR spekter[32] s vysokým rozlišením. Může sloužit i pro zobrazení a úpravu skutečných dat, která už ale prošla Fourierovou transformací, WINDNMR neumí pracovat s FIDy. Všechna spektra mohou být uložena i jako obrázky nabo vyexportována do jiných aplikací.
Nechemické programy
[a] JRE lze zdarma získat např. na adrese http://java.com/en/download/manual.jsp, k dispozici je buď automatická instalace bez nutnosti cokoli spouštět a nebo stažení samotného instalačního souboru [b]
[c] návod a soubor příkazů je např. na adrese http://www.baldridge.unizh.ch/education/gamess/input.html [d] DFT metody výpočtu jsou metody, které počítají pouze s elektronovou hustotou na atomech [e] Ke správnému chodu programu Numerical Chameleon je třeba mít mít nainstalované JRE. Instalační soubor programu NumericalChameleon (nc-install.jar) lze pak v operačním systému Windows spustit běžným způsobem, tzn. dvojím kliknutím, ovšem někdy se stane že přípona JAR už byla dříve "ukradena" jiným programem. V tomto případě je ještě nutné buď manuálně přiřadit tuto příponu správnému programu (tedy souboru javaw.exe, který je součástí JRE) a nebo jednodušeji, spustit ještě před samotnou instalací aplikaci jarfix.exe, která toto provede automaticky. [1]ACS Style Guide: Examples for citing references. http://chemistry.library.wisc.edu/instruction/acstyle.htm#internet (dne 05/06/06) [2] Open Source. http://www.opensource.org (dne 20/12/05) [3] SourceForge.net: Compile Farm. http://sourceforge.net/docs/compile_farm (26/04/06) [4] ACD/Labs. http://acdlabs.com (dne 25/12/05) [5] Elsevier MDL. http://mdl.com (dne 25/12/05) [6] CambridgeSoft. http://cambridgesoft.com (dne 25/12/05) [7] Free Software and science. http://fsffrance.org/science (dne 19/04/06) The Free Software Foundation. http://www.fsf.org (dne 29/01/06) Software&Tools. http://ncrr.pnl.gov/software/ (dne 09/04/06) Finnish IT Center for Science. http://www.csc.fi/chem (dne 17/03/06) National HPCC Software Exchange. http://www.nhse.org (dne 18/04/06) Netscape Browser Archive. http://dir.sillydog.org (dne 18/04/06) International Union of Crystallography. http://www.iucr.org (dne 17/03/06) Free Download Manager. http://www.freedownloadmanager.org (dne 17/03/06) [8] The Open Source Definition. http://opensource.org/docs/definition.php (dne 20/12/05) [9] GNU Public Licence. http://www.gnu.org/licenses/gpl.txt (dne 25/12/05) [10] SourceForge. http://sourceforge.net (dne 02/02/06) [11]The Blue Obelisk Movement. http://blueobelisk.org (dne 18/04/06) Eclipse Community. http://www.eclipse.org (dne 18/04/06) The Open Science Project. http://www.openscience.org (dne 19/04/06) Chemoinformatics. http://www.cheminformatics.org (dne 18/04/06) The Open Directory project. http://www.dmoz.org (dne 20/04/06) Free downloads Encyclopedia. http://softpedia.com (dne 12/05/06) TheOpenCD. Collection. http://theopencd.sunsite.dk (dne 04/06/06) [12] Java. http://java.sun.com (dne 20/12/05) [13] The JavaTM Tutorial. http://java.sun.com/docs/books/tutorial/deployment/jar (dne 29/05/06) [14] Jar File Overview. http://java.sun.com/j2se/1.4.2/docs/guide/jar/jarGuide.html (dne 29/05/06) [15] Chemistry Java Applets. http://www.101science.com/chemJAVA.htm (dne 02/02/06) Application of Java to Chemistry. http://www.ch.ic.ac.uk/java (dne 02/02/06) Educational Applets. http://www.chem.uci.edu/undergrad/applets (dne 02/02/06) [16] The Python Programming Language. http://python.org (dne 27/01/05) [17] Computational Chemistry Software. http://bogense.chem.sdu.dk/~icc/software.html (dne 21/04/06) Computational Chemistry List. http://www.ccl.net/chemistry/links/software (dne 24/03/06) [18] Computational Chemistry: Basic terms. http://www.chemistryexplained.com/Co-Di/Computational-Chemistry.html (dne 05/06/06) [19] Quantum Chemistry. http://www.baldridge.unizh.ch/education/quantum.html (dne 18/04/06) [20] CML. http://cml.sourceforge.net (dne 13/04/05) [21] Chemical Informations Systems. http://daylight.com (dne 01/06/05) [22] IUPAC:InChI. www.iupac.org/inchi (dne 01/02/06) [23] Protein Data Bank. http://www.rcsb.org (dne 02/03/06) [24] ExPASy Proteomics Server. http://www.expasy.ch (dne 05/04/06) [25] PCGamess. http://www.scl.ameslab.gov/gamess/register (dne 12/3/06) [26] Gaussian Input Overview. http://www.gaussian.com/g_ur/m_input.htm (dne 02/06/06) [27] MOPAC7 Download. http://sourceforge.net/projects/mopac7 (dne 25/05/06) [28] The MathWorks. http://www.mathworks.com (dne 20/11/05) [29] Molecular Mass Calculator. http://medlib.med.utah.edu/masspec/mole.htm (dne 02/04/06) Mass Spectroscopy Utilities. http://www.chemistry-software.com/masspec.htm (dne 02/04/06) Mass Spectrometry Software from SIS. http://www.sisweb.com/software.htm (dne 02/04/06) [30] IUPAC Provisional Recommendations for the construction and use of ligand abbreviations . http://www.iupac.org/reports/provisional/abstract04/RB-prs310804/TableVII-3.04.pdf (dne 05/06/06) [31] MestRe-S Registration. http://www.mestrec.com/register.php (dne 12/04/06) [32] NMR Related Software. http://www.spincore.com/nmrinfo (dne 06/01/06) NMRShift Database. http://www.nmrshiftdb.org (dne 07/03/06) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||










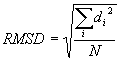 , kde N je po
, kde N je po